Discovery of Selective EGFR Inhibitors against L858R/T790M Resistance Mutation
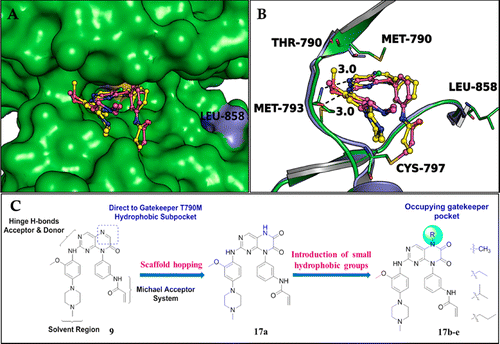
Starting with a previously reported irreversible EGFR inhibitor, we developed 6,7-dioxo-6,7-dihydropteridine-based selective and cell-active inhibitors of EGFRL858R/T790M mutants. Analysis of the crystal structure of EGFRT790M and EGFRWT suggested that introduction of substituents at the N-5 position of the dihydropteridine scaffold would lead to an increase in both EGFRL858R/T790M activity and selectivity because these can provide suitable vectors toward the gatekeeper Met790 pocket. While in the EGFRWT kinase, the smaller and more polar threonine residue would reduce the hydrophobic interaction between the gatekeeper residue and the 5-subsitutent, leading to lower affinity. As expected, introduction of hydrophobic groups, such as an isopropyl (17d) at the 5-position, led to an increase in EGFRL858R/T790M inhibitory activity, which agrees well with our design hypothesis. Importantly, 17d displayed strong antiproliferation activities on H1975 cancer cells, while its effect on the A431 cancer cells was significantly less potent. A further in vivo antitumor efficacy evaluation on 17d suggested that it significantly inhibited the tumor growth in H1975 NSCLC xenograft mouse model by po dosing at 50 mg kg–1d–1 for 14 days. The encouraging selectivity and in vivo antitumor effects of compound 17dsupported that it might be used as a promising lead compound for further development as a selective EGFR inhibitor to overcome the EGFR resistance mutations.
J. Med. Chem., 2016. 59 : 7111-7124
DOI: 10.1021/acs.jmedchem.6b00403
User Login
Web Services
Contact us
Honglin Li's Lab
Shanghai Key Laboratory of New Drug Design
School of Pharmacy
East China University of Sci. & Tech.
Room 527, Building 18, 130 Meilong Road,
Shanghai, 200237, P. R. China
Tel: (86) 21 6425 0213 Prof. Honglin Li
hlli@ecust.edu.cn
Copyright © 2026 Prof. HongLin Li's Group, School of Pharmacy, East China University of Science & Technology · All Right Reserved.
沪ICP备19004698号-1 |  沪公网安备31011302004713号
沪公网安备31011302004713号