News
scRISE:基于AI的单细胞聚类算法
2024-06-07华东理工大学/华东师范大学药学院的李诗良/李洪林教授团队在国际知名学术期刊Oncogene上发表了题为"Clustering single-cell RNA sequencing data via iterative smoothing and self-supervised discriminative embedding"的研究论文。这项研究提出了一种创新的深度学习聚类方法——scRISE模型,为单细胞RNA测序 (scRNA-seq) 数据的聚类分析提供了高效的解决方案。scRISE模型是一种巧妙地融合了迭代平滑技术和自监督判别嵌入技术的深度学习框架。该模型的架构由两个主要模块构成:迭代平滑模块和自监督判别嵌入模块,以协同优化单细胞数据的聚类分析过程。
单细胞RNA测序 (scRNA-seq) 技术是现代生物学研究的一大突破,它允许研究人员在单个细胞层面上测量基因表达,为我们理解细胞多样性和复杂生物系统的细胞组成提供了前所未有的视角。在scRNA-seq数据分析中,聚类分析是识别细胞类型和亚群的一个关键步骤,这要求我们要有精确的相似性度量方法和有效的算法来应对这些数据的复杂性。为了提高聚类分析的准确性,研究团队开发了一种名为scRISE的新型深度学习聚类方法。scRISE模型的创新之处在于两个主要方面:
- 迭代平滑技术:scRISE采用了拉普拉斯平滑技术,通过迭代过程逐步优化数据,有效减少了高频噪声,提高了数据质量。
- 自监督学习:模型中集成的自监督判别嵌入模块,能够自动选择正负样本对,减少了人为干预,提高了聚类的准确性和效率。
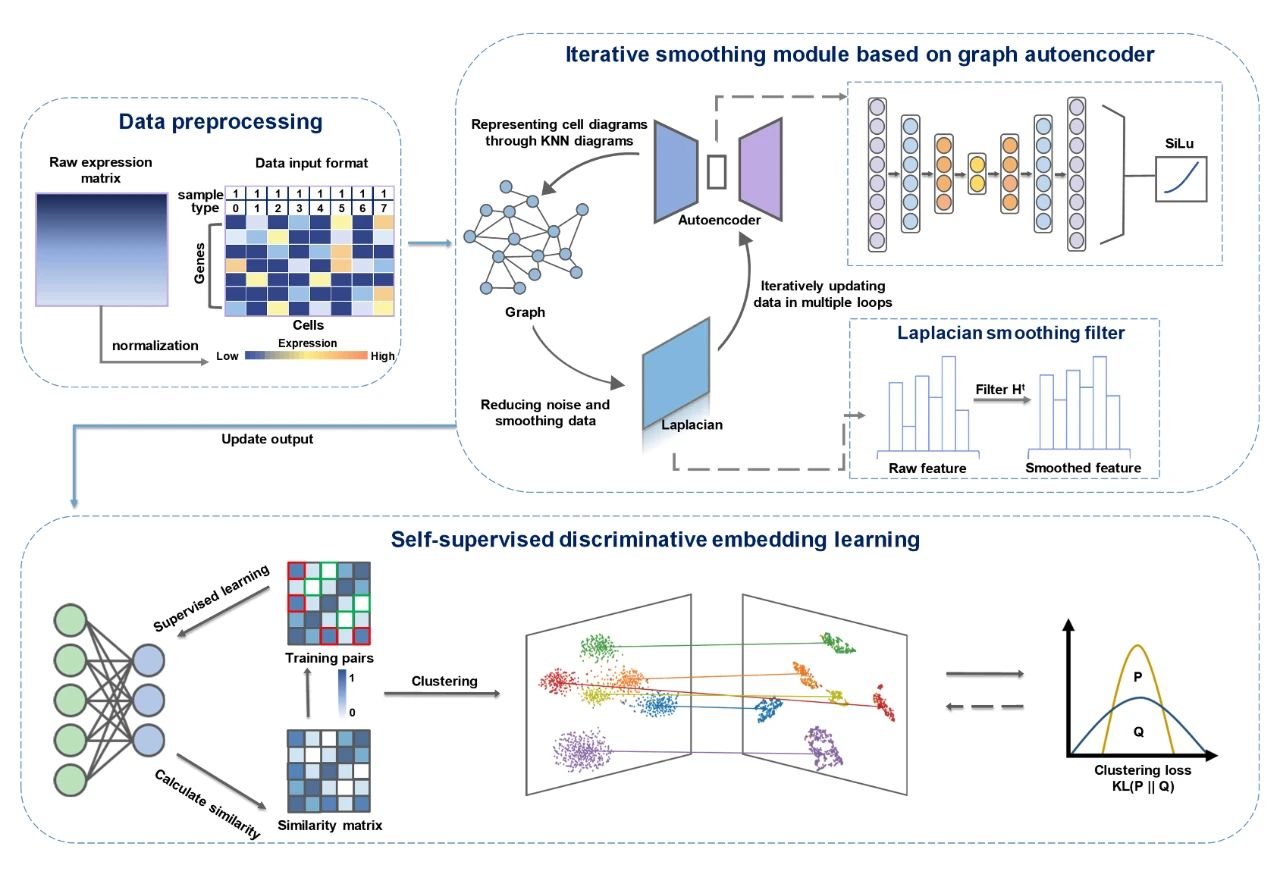
scRISE方法由两个主要模块构成,如图1所示。首先,研究团队使用了一个基于图自编码器的迭代平滑模块,该模块结合了自编码器和拉普拉斯平滑滤波器来平滑和重建可能存在噪声、不完整或粗糙的数据,同时融入了细胞间的结构信息。自编码器能够准确地重建数据中的主要信号,而拉普拉斯平滑滤波器通过平滑处理进一步改善数据质量,减少噪声的影响。这个迭代过程不断地更新重建数据,逐步提高单细胞数据的准确性和稳定性。接着,采用了一个自监督判别嵌入模块,该模块利用细胞间的相似性来选择正负样本对,以确定数据中固有的簇。在这个模块中,正负样本对的阈值是自适应选择的,这样属于同一簇的样本自然会被推到一起,而来自不同簇的样本则在嵌入空间中相互排斥。该模块旨在通过学习数据分布中固有的相似性结构来增强聚类性能。通过这两个模块的融合,scRISE有效地去除了数据中的不兼容和噪声信号,并实现了无需大量人工干预的自监督聚类。
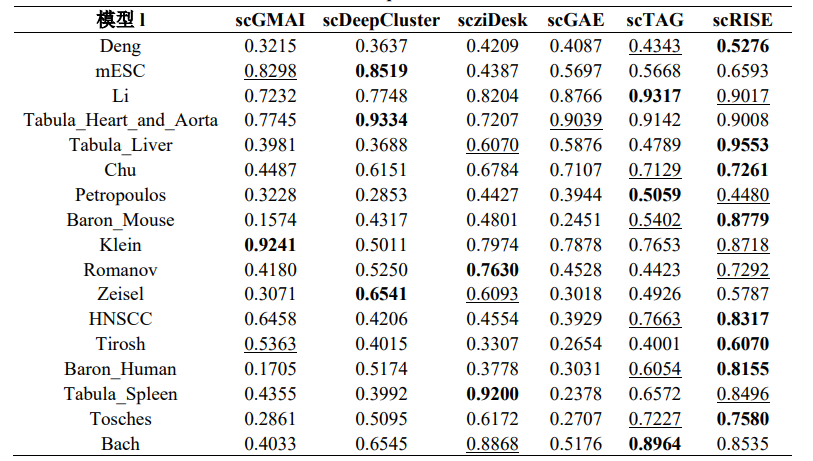
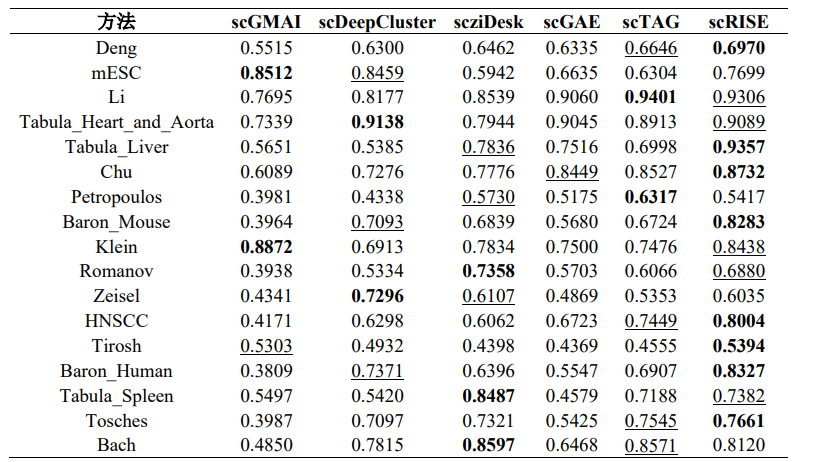
在17个公开的真实scRNA-seq数据集上,将scRISE与其他五种深度学习聚类方法进行了比较,如表1和表2所示,scRISE在所有数据集上的ARI和NMI值均排名第一或第二,明显优于其他方法。scRISE在Baron_Human数据集上表现出色,其ARI和NMI值分别达到了0.8155和0.8327,证明了其在区分不同细胞类型方面的高准确性。在对scRISE模型的消融研究中(图2),数据平滑任务显著提升了聚类的准确性和精确度,并在所有测试数据集中显著提高了聚类精度。聚类模块进一步提高了聚类精度,但在其他数据集中(如Li, Spleen, Bach),当数据平滑任务已经明显改善聚类精度时,聚类模块的增益有限。
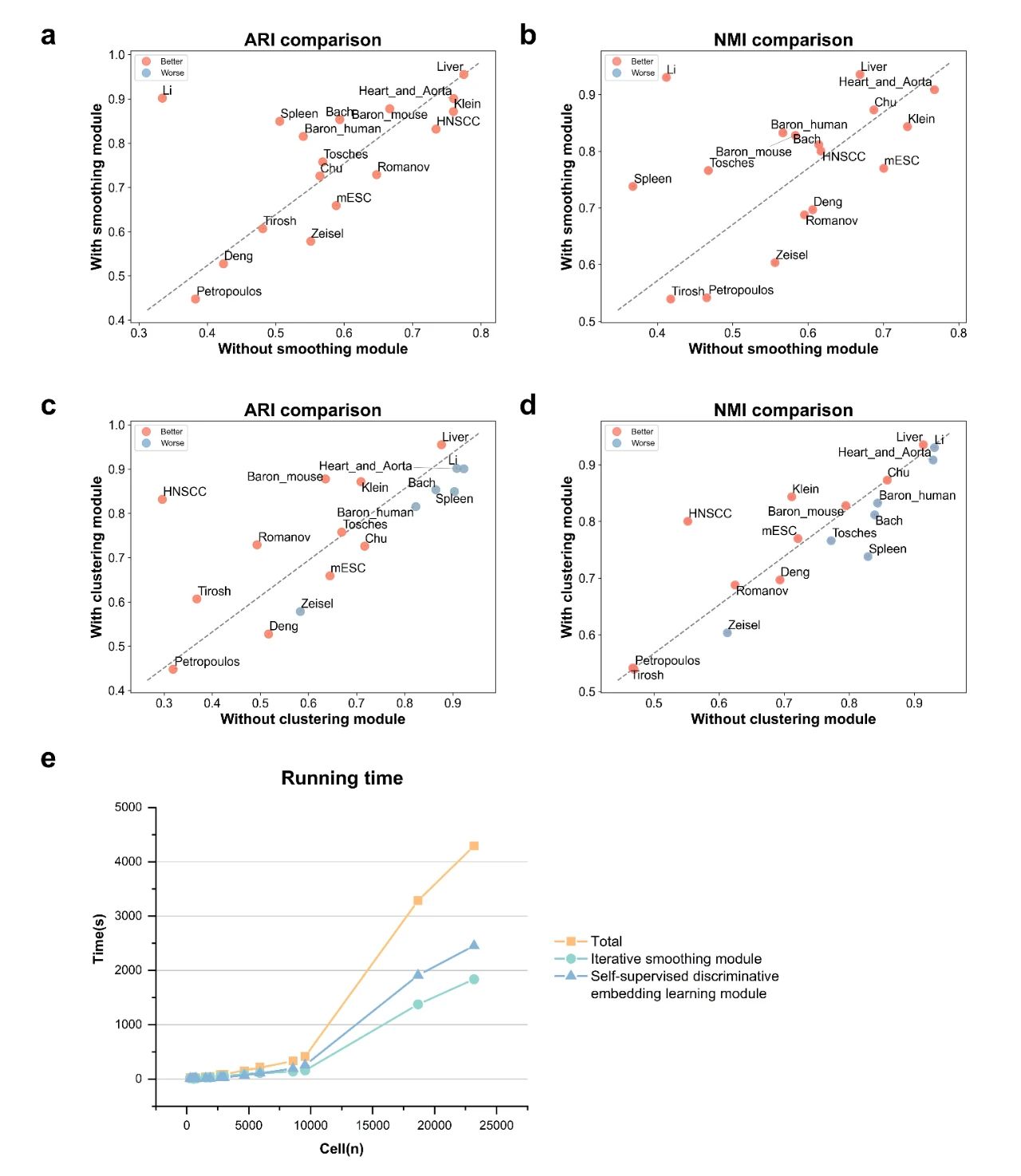
消融研究的结果表明,数据平滑任务有效滤除了数据中的高频噪声,而聚类模块则通过考虑细胞间的相似性和捕获区分性表达模式来提升聚类性能。为了评估scRISE的可扩展性,研究团队在17个真实scRNA-seq数据集上测试了算法的运行时间。测试内容包括scRISE模型的总运行时间,以及其核心模块——基于自编码器的迭代平滑模块和自适应编码器聚类模块的运行时间。结果显示,scRISE具有良好的可扩展性,其运行时间与数据集的规模呈类二次方的关系,这意味着即使在面对大规模数据集时,scRISE也能保持合理的运行时间,不会显著增加计算成本。
综上所述,scRISE模型在单细胞RNA测序数据聚类方面展现出优异的性能,它不仅能够提供精确且具有生物学意义的聚类结果,还表现出了良好的泛化能力和可扩展性。scRISE模型作为一个功能强大的工具,将极大地促进单细胞生物学研究的深入发展,为探索细胞异质性和细胞状态的动态变化提供新的视角和方法。
eTSN: 疾病—靶标知识图谱可视化平台
2022-11-09华东理工大学药学院上海市新药设计重点实验李诗良/李洪林团队在Briefings in Bioinformatics上发表了文章e-TSN: an interactive visual exploration platform for target-disease knowledge mapping from literature,开发了目前最大的基于生物医学文本挖掘的可视化疾病—靶标知识图谱的多功能平台(e-TSN: the explorer for Target Significance and Novelty, http://www.lilab-ecust.cn/etsn/)。该平台对200多万篇生物医学全文文献中疾病-靶标关系数据进行提取,生成与药物-靶标-疾病相关的知识图谱,通过可视化表示,可为疾病提供潜在靶标谱,为靶标提供疾病谱,并关联已有和在研药物数据库,为寻找合适的靶点这一难点问题提供了实用的解决方案,同时也为数据驱动的靶点临床决策和新药研发立项提供了线索和理论依据。
研究背景
新药研发是一项耗时长、投资大、风险高的系统工程,平均耗资高达26亿美元,耗时10年之久。药物靶标发现作为新药研发的源头,对药物研发的成功率起到决定性作用。基于“新靶点、新机制”的药物发现已经成为现代药物开发的主流,对靶标和疾病、药物相互作用知识的了解对于开发新药和药物再利用至关重要,不仅可以促进人们在分子水平上对药物作用的理解,而且还有助于提高药物开发效率。据统计,全世界已知的疾病大约有3万种,人类基因/靶蛋白数量多达2万多种,但在已批准上市的药物中,涉及的靶标数量仅占人类蛋白的10%左右,大多针对肿瘤学、传染病、神经病学、免疫学和呼吸系统等治疗领域。因此,仍有一大部分治疗领域存在未被满足的临床需求,为其寻找新的治疗靶点和药物迫在眉睫。对于制药企业来说,寻找新的治疗领域或新靶点开展新的研发管线也十分重要。
研究人员在对感兴趣的疾病、靶标进行研究时离不开对相关文献知识的调研。然而,随着生物医学的快速发展,科学文献正以指数级的速度迅速增加,这极大地推动了靶标发现和识别过程,为科学家提供了寻找疾病-靶标相关性的机会。生物医学文献的显著增长使得科学家比以往任何时候都更难找到和吸收所有与他们的研究相关的文献,即便是业内的行家也无法完全依赖于传统的人工检索方式从中凝练出生物医学知识。
因此,如何从海量文本数据中自动、高效读取有价值信息成为有效获取靶标-疾病知识的关键。尽管目前已有多种文本挖掘算法用于自动从文本中提取关系,但在将这些信息与现有数据库相联系并转化为用户可以理解的知识之间仍存在很大的滞后。知识图谱(KGs)利用强大的算法系统地填补了靶标-疾病关系的未知区域,并对产生疾病的基因和机制提供了新的见解,可以为药物新靶标发现提供信息技术支撑。如何从海量非结构化的文本数据中提取潜在疾病-靶标相互作用信息;如何将从文本中提取的知识与现有数据库的知识相融合;如何可视化知识图谱以帮助用户研究见解是亟需解决的关键问题。
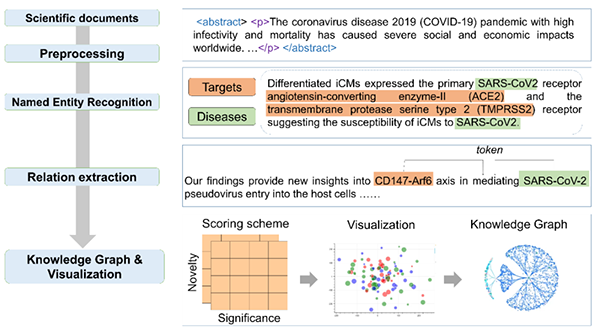
图1 靶标-疾病知识图谱的交互式可视化平台e-TSN搭建流程
研究内容
作者开发的系统框架具体流程如图1所示。首先构建疾病、靶标实体名称组成的词典:疾病名称覆盖感染性疾病(Disease by infectious agent)、组织结构实体疾病(Disease of anatomical entity)、细胞增殖病(Disease of cellular proliferation)、代谢疾病(Disease of metabolism)、精神疾病(Disease of mental health)、遗传病(genetic disease)、身体紊乱(physical disorder)、综合征(syndrome)、罕见病(rare disease)共9类疾病,靶标名称涵盖酶(Enzyme)、表观遗传因子(Epigenetic)、G蛋白偶联受体(GPCR)、孤儿G蛋白偶联受体(oGPCR)、离子通道(Ion channel)、激酶(Kinase)、核受体(Nuclear receptor)、转录因子(Transcription factor)、转运蛋白(Transporter)以及尚未明确分类的靶标(Non-IDG)共10类蛋白家族。接着对获取的生物医学全文文献进行预处理;通过采用基于自然语言处理的命名实体识别和关系抽取技术对PubMed Central数据库中超过200多万篇生物医学全文文献中疾病-靶标关系数据进行提取。为了从数百万个文本中集成靶标和疾病的关系,作者首先定义了两个新型指标:1)重要性:用于衡量靶标和疾病两个实体之间的关联程度;2)新颖性:用于表示靶标被研究或未被研究的程度。通过构建新颖的基于文献统计学的重要性与新颖性评分方法加权整合了靶标和疾病的关联数据,并将其与先验的关系数据进行集成,构建了目前最大的疾病-靶标关系数据库,包含超过1.7万种疾病与大于2万种基因/蛋白质之间3亿多条潜在的关系;进一步整合DrugBank数据库中靶标与上市、在研药物关系、ChEMBL数据库中活性分子关系数据,构建疾病-靶标-化合物实体之间的关系网络,最终将关系数据通过网页可视化平台呈现以帮助研究者进行快捷的知识查询和探索。
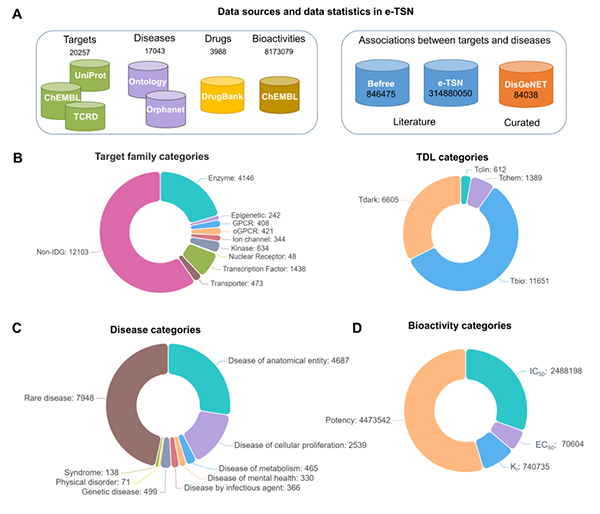
图2 e-TSN数据库统计分析
平台功能
e-TSN是一个可用于调查和分析靶标-疾病复杂网络的可视化工具,有助于研究人员了解疾病表型的潜在机制,提高药物的发现和开发效率,特别是当面对突发的传染病大流行如新冠、流感等紧急情况,其能为迅速发现有效药和特效药提供科学线索和技术平台支撑。1)用户可以通过检索感兴趣疾病的关键词(例如“SARS-CoV-2”),获得所有相关靶标的重要性和新颖性分布(图3),以帮助用户快速对潜在靶标进行优先级排序并选择最感兴趣的开展研究;同时,用户可以通过点击散点图面板上显示的任一靶标名称获得详细的注释信息,包括已批准和在研的药物和相关的生物活性分子,为靶标发现提供更充分的考量(图5)。2)反之,用户还可以探索与特定靶标相关疾病的重要性与新颖性分布(例如“ACE2”),以寻找与感兴趣的靶基因相关的其他适应症并进行药物重定位(图4)。
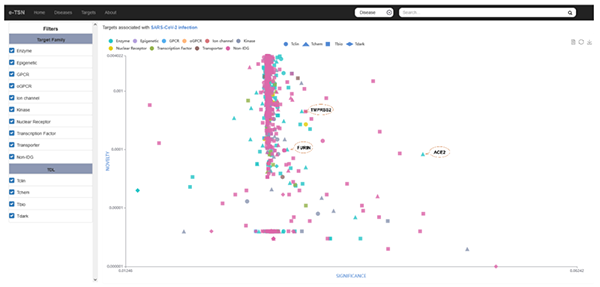
图3 e-TSN界面:与SARS-CoV-2相关靶点重要性与新颖性分布
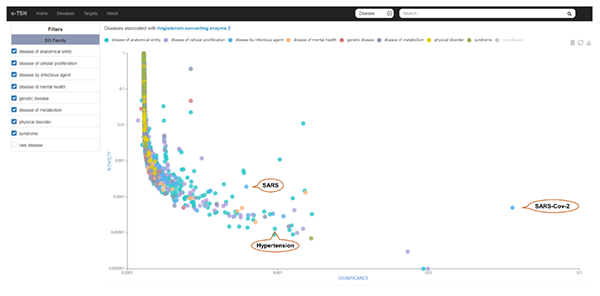
图4 e-TSN界面:与ACE2相关疾病重要性与新颖性分布

图5 e-TSN提供有关靶标和疾病的全面信息
研究总结
综上所述,该团队提出了一种基于文献的知识合成和发现的新方法,构建疾病-靶标相互作用知识图谱,建立了一个交互式可视化平台e-TSN,通过知识图谱来捕获和表示疾病与靶标之间的关系,极大填补了靶标-疾病关系的未知区域,有助于理解疾病机理和表型,并结合现有的靶标-药物关系数据,对特定疾病新靶点选择、新药设计以及药物重定位具有较好的指导意义,可为现有的AI药物设计提供思路和指明方向,有助于进一步缩短新药从立项到临床周期,从而提高新药研发效率。
该工作的第一作者是华东理工大学的博士研究生冯紫燕同学,通讯作者为华东理工大学药学院李诗良副教授。相关算法和软件已申请软件著作权保护,该研究项目得到了国家自然科学基金的资助。
选择性JAK2抑制剂
2022-10-26上海市新药设计重点实验室李洪林教授课题组设计改造的新型选择性JAK2抑制剂对骨髓增殖性肿瘤有显著治疗效果,研究结果“Efficacy of WWQ-131, a highly selective JAK2 inhibitor, in mouse models of myeloproliferative neoplasms”在《Biomedicine & Pharmacotherapy》上发表(Biomedicine & Pharmacotherapy., 2022, 156, 113884)。
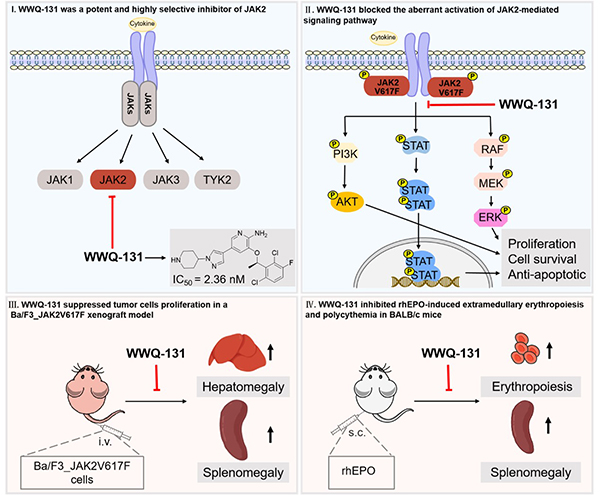
骨髓增生性肿瘤(MPN)是一类恶性髓系肿瘤,其特征是骨髓中过度产生终末分化成熟血细胞。MPN患者有一系列临床表现,从血红蛋白/红细胞比容和血小板计数升高到进行性细胞减少、骨髓纤维化和转化为急性髓性白血病(AML)的风险,伴有典型的临床特征脾肿大和严重的体质症状(疲劳、恶病质、瘙痒和骨痛)。异基因造血干细胞移植(AHSCT)治疗是骨髓增生症(MF)患者的唯一治疗选择。然而,由于高发病率和死亡率,AHSCT受到严格限制。Janus激酶2(JAK2)信号通路的过度激活导致骨髓增生性肿瘤(MPNs),靶向JAK2可作为治疗MPNs的有效策略。
本研究是针对课题组前期设计改造得到的高选择性JAK2抑制剂12k的后续研究。这里,我们报道了WWQ-131(12k的R对映体)是比外消旋体12k更有效的试剂,WWQ-131对JAK2的抑制活性IC50为2.36nM,对JAK2的选择性分别比JAK1和JAK3高182和171倍。在JAK2V617F依赖性细胞系中,WWQ-131有效抑制细胞增殖,诱导细胞周期阻滞在G2/M期和凋亡,并阻断JAK2信号通路的异常激活。在Ba/F3_JAK2V617F驱动的小鼠疾病模型中,WWQ-131有效抑制脾脏和肝脏中STAT5磷酸化,并抑制Ba/F3_ JAK2V17F细胞在体内的扩散和增殖。此外,WWQ-131抑制rhEPO诱导的小鼠髓外红细胞生成和红细胞增多症,以及红细胞比容和脾脏大小,尤其对白细胞计数没有影响。此外,在这两种MPN模型中,WWQ-131(75 mg/kg)表现出比Fedrratinib(120 mg/kg)更强的治疗效果。综上所述,这项研究表明WWQ-131将是治疗MPN的一个有前途的候选药物。
该研究项目得到了自然基金委、科技部及上海市科委的资助。
原文链接:https://www.sciencedirect.com/science/article/pii/S0753332222012732
CIRS: 快速高效的Markush结构图像识别系统
2022-10-21华东理工大学药学院上海市新药设计重点实验/华东师范大学人工智能新药创智中心李洪林/张凯团队在Briefings in Bioinformatics上发表题为Multi-Modal Chemical Information Reconstruction from Images and Texts for Exploring the Near-Drug Space的文章。研究团队历时近4年之久,基于多模态学习发展了一种从文本和图像中进行化学信息重建系统CIRS(Chemical Information Reconstruction System),实现快速高效的Markush结构识别以及其与可变取代基文本的信息重建任务,进而自动提取化学专利中的化学分子结构。该方法是目前唯一一个快速高效的Markush结构图像识别系统,对“近药空间“的构建以及候选新药的设计具有重要意义。
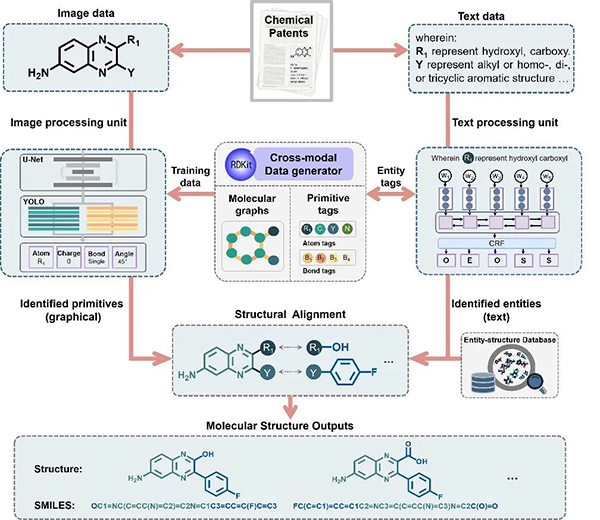
图1 CIRS 框架
研究背景
发现具有优良生物学效应的新化学实体是新药发现的目的及核心问题。因此,多样性化学空间的探索,特别是类药空间(Drug-like Space)的探索是化学信息学家和药物化学家最关注的技术难点问题。目前可合成的化学真实空间(Real Space)已达到1010(百亿)的级别,而科学家初步估算的类药化学空间可能包含1060符合类药五规则的分子。传统的分子虚拟空间构建方法包括枚举法和虚拟库等方法,由于生成的分子多样性、可合成性及成药性不足,上述方法愈发无法满足需求了,随着近年来人工智能(AI)在药物设计中的涉入和计算能力的大幅提升,使得更大化学空间的构建成为可能。但是否构建的化学空间越大越好?答案当然不是!从巨大的化学空间中快速识别出包含活性分子的区域即药效空间(Pharmacological space)才是药物发现的关键所在。随着网络技术和信息技术的快速发展,文献、专利、网页、图片、生化数据库等各类知识为化学空间和知识图谱构建提供了各种信息来源,其中,专利类文献在制药业和生物技术中始终占据重要地位,这不仅是因为其具有信息披露早、数据覆盖面全、数据来源可靠的特点,还因为具有可合成性质和针对特定靶点的主要活性候选分子仅在专利中公开,而散布在这些药物专利的核心结构周围的药效空间,是具有最大可能发展成为候选药物的 “近药空间”分子(Near-Drug Space)。不同于普通文献数据来源,专利文献往往是以一个类属(Genus)化合物发明的通式——马库什(Markush)结构扩大其广泛的保护范围,这在化学领域——特别是化学药物领域尤为重要。因具有极强概括能力,Markush这一独特的结构在化学和生物医药领域被广泛应用。Markush概念实质上是一种简化和概括,以马库什方式撰写的文献和专利权利要求实质上是用简化的方法来描述具有相同或者相似功能的一类结构、设计或者系统,而这种简化方法却提供了广泛的法律覆盖范围,因此Markush结构也是生物医药专利研究之热点和专利纠纷主要集中点。尽管以Markush结构为核心部分,通过组合可变取代基能够衍生出大量性质相似的“近药空间”分子,进而为新药研发提供优质的起点分子。但其可变取代基的复杂性使得马库什结构的检索和识别成为化学信息学领域的一大难题,亦是化学信息学领域几十年来的研究热点。同时,专利文献中Markush结构的绘图风格(原子标签、键描绘风格等)不规范、开放访问数据集匮乏以及传统算法效率较低等问题限制了Markush结构识别相关研究的发展。现有的分子结构识别软件也只是简单的图像分子识别,多不能解决Markush结构中功能基团和R基团的识别,类如image2smiles也仅能识别简单的R基团却不支持特殊的键形式。因此,开发快速高效的Markush结构识别工具,既可以提高化学分子结构数据识别效率以解决该领域难点技术问题,也可为“近药空间”的扩展提供广阔数据来源,进而提高药物发现成功率并降低药物研发成本。
CIRS 系统
因专利类文献中涵盖不同形式的化学信息,挖掘不同领域知识之间的联系对提取更为准确的化学信息至关重要。尤其Markush结构图像和可变取代基实体文本具有高度异构性,如何快速高效地融合两个领域知识并完成信息自动提取是化学信息领域的关键挑战之一。CIRS设计了图像处理单元(左)、异构数据生成器(中)和文本处理单元(右)(图1),可用于同时处理专利文献中的Markush结构图像和可变取代基文本并通过二者内在关联规则完成化学信息的重建。
系统框架具体流程如下:首先,数据生成器将生成Markush结构图像和原子/键标签(像素级),然后将其作为训练数据输入到图像处理单元。在图像处理单元中使用了图像分割模块(U-Net3+)和目标分类模块(YOLO),以便将图像数据中的像素分割成原子、键和电荷并为其分配正确的标签。在右侧的文本处理单元中,采用BiLSTM-CRF模型的完成化学实体识别,以识别文本中的化学实体,识别出的实体则通过建立的结构数据库转化为SMILES格式,最后将这两部分的输出进行融合,通过左侧的原子标签和右侧的实体类型进行化学信息重建,组合出图像及文本中涵盖的化学实体结构。值得一提的是,作为中心模块的数据生成器对生成训练样本的数量和多样性没有严格限制,它可以根据用户需求随机修改分子,因此为图像处理和文本处理单元的泛化性能提供基础,这也是CIRS适用于从大量知识来源中提取化学信息并推广到各类文献中分子结构提取的关键。
在Markush结构图像识别方面,该团队基于异构数据生成器,随机模拟出含有官能团、官能团占位符(R 基)和椒盐噪声等的分子图,进而提高模型的泛化能力。图2显示了生成的分子图像的几个示例,图像主要包含R-基团、官能团、环R键和随机椒盐噪声。
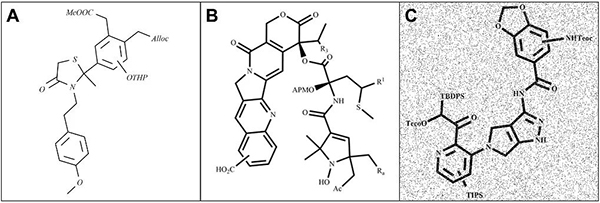
图2 生成的分子图像实例
进一步通过先进算法组合,即语义分割模块(U-Net3+)和分类模块(YOLO)(图3A),完善Markush结构图像识别准确率,其中对分子图像中原子、键型电荷的识别精确度超过98%,同时对Markush结构图像的识别正确率超过97%(图3B)。
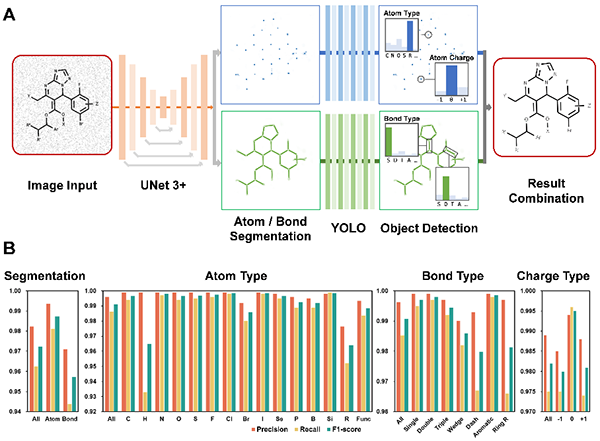
图3 CIRS中图像处理单元的语义分割及分类模型(A)及评估结果(B)
此外,识别文本中的化学实体并将其转换为预定义的标签是实现不同领域化学信息融合的基础。该团队通过手工标注克服文本识别训练集匮乏难题,并通过数据增强技术扩充标注数据规模(图4A),采用经典的BiLSTM-CRF模型完成文本描述中的实体识别(图4B),实现Markush结构图像与可变取代基文本描述这两个不同领域化学信息融合,基本解决了取代基文本直接转化为可编辑的结构数据的难点问题(准确率大于97%)(图4C)。
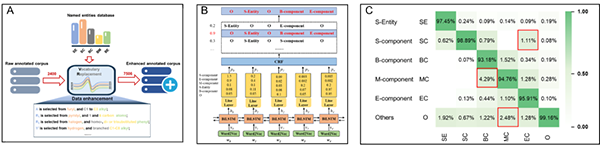
图4 文本信息处理单元中的数据增强规则(A)、模型框架(B)以及模型表现(C)
同时,为了解决文本数据无法转化为结构数据问题,该团队建立了包含取代基结构信息的结构化数据库,实现化学文本直接转换为药物化学家可编辑的化学结构,进而有效地用于构建可扩展的分子结构,从而实现以Markush结构为中心快速构建“近药空间”的分子。
最后,为了诠释CIRS能够实现不同领域知识重建并具有在现实场景中进行自动信息提取与重建的潜力,该团队进行了实际案例研究(图5),通过CIRS系统处理专利中的Markush结构图像和取代基实体文本,可获得大量的分子结构来促进近药物分子的生成,并有望为药物化学家构建一个具有重要意义的近药空间。此外,从化学专利中自动提取信息可以更容易地定义专利覆盖范围,从而避免未来药物发现中的化学知识产权冲突。

图5 CIRS从化学专利中自动提取结构实例研究
综上所述,团队提出了一个多模态化学信息重建系统CIRS,该系统实现了快速高效的马库什结构识别,同时可以自动识别文本中化学实体并转化为结构数据,并通过多模态策略学习信息融合规则,最终实现不同领域知识的化学信息重建。CIRS在探索“近药空间”领域是一个极具潜力的化学信息重建工具,可为“近药分子”生成与优化等任务提供高质量的数据支持。基于CIRS系统,团队现已开展药物专利核心数据库和知识库(如知识图谱)的建设,为实现化学知识的智能检索和近药空间药物设计奠定研究基础。
该工作的共同第一作者是华东理工大学的博士研究生王洁和沈子豪同学,相关算法和软件已申请专利和软件著作权保护。该成果得到了国家自然科学基金等项目的支持。
VRPharmer 国内首个可交互式计算的VR药物设计软件
2022-09-23目前,基于药效团的虚拟筛选(virtual screening, VS)已作为一类快速且有效的计算机辅助药物设计技术,参与到药物研发流程中。同时,VR(Virtual Reality,VR)以多感知性、沉浸性、交互性和构想性为特征被广泛应用,有助于真实感知微观分子世界。因此,需要将高性能科学计算和VR技术相结合开发药效团虚拟筛选平台,但目前国内拥有自主知识产权的此类软件甚微,无法为国内新药研发做强有力的软硬件支撑。
Oxford University Press网站报道了华东理工大学药学院/华东师范大学人工智能新药创智中心李洪林教授课题组和华东师范大学计算机学院何高奇副教授课题组题为“VRPharmer: Bringing Virtual Reality into Pharmacophore-based Virtual Screening with Interactive Exploration and Realistic Visualization”的最新成果。该成果发表于国际著名学术期刊Bioinformatics。研究团队将科学计算模型、VR与药物设计相结合,首次发布了用户友好、兼具科学性、艺术性与趣味性的基于药效团虚拟筛选全流程交互式可视计算软件VRPharmer。
积科学研究之跬步,至创新成果以千里
在计算机辅助药物设计(CADD)中,由于药效团筛选能快速、准确地发现先导化合物,得到工业界和学术界的广泛应用,也成为人工智能药物设计中最为核心的环节[2]。华东理工大学李洪林教授团队在过去十几年间致力于发展原创精准的靶标和药物发现方法,秉持“做自己的软件”、“做实用的软件”的初衷,相继开发了利用反向药效团匹配方法识别探针小分子(药物、天然产物、或其他靶标不明确的新化合物)潜在靶标的在线预测平台:PharmMapper、基于药效团虚拟筛选的图形化药物设计软件ePharmer等,全球科研用户超3.5万,获2020年教育部自然科学一等奖。厚积而薄发,VRPharmer正是在前期工作基础上的又一次创新和突破。
构建真实感三维虚拟空间,元宇宙里探索药物设计
在日益复杂的生物大分子和药物分子的设计中,研究对象结构复杂、功能多样、尺度微观,利用传统的输入设备对化合物分子进行交互操作时,面临精度不足和相互遮挡等限制,无法精确感知深度信息和相对距离,也很难理解基于药效团虚拟筛选中结构相似性和特征相似性的原理。
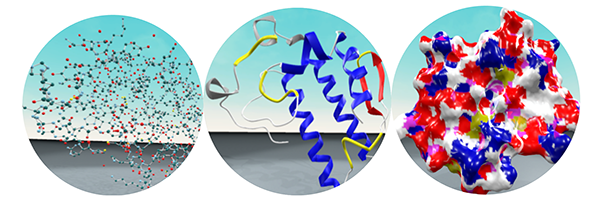
图1 多尺度分子显示。一级结构(左);二级结构(中);表面结构(右)
VRPharmer通过先进元宇宙的VR交互技术,构建真实感三维虚拟空间,结合游戏过程的交互模式和视觉素材的美学设计,利用2D/2.5D/3D渲染技术表达分子结构,使科研人员脱离2D屏幕、鼠标和键盘的束缚,身临其境于分子内部,零距离、全方位、多角度、沉浸式进行分子编辑,感受基于药效团的药物设计的过程。
支持多模操作和数据分析,高效完成科学计算
VR价值不仅限于体验,更在于交互计算功能。基于团队前期发展的PharmMapper和ePharmer,系统设计了四个关键环节及相应的控制菜单:Load(本地或远程加载分子文件);Visualization(查看分子结构);Render(渲染分子模型);Screening(分子虚拟筛选)。Screening模块筛选出一批具有潜在生物活性的小分子并确定每个分子与靶标蛋白的结合模式。在科学计算过程中,系统设计了两个独特的交互式筛选模式:评分模式和最优化模式。
- 评分模式计算蛋白质和小分子的匹配得分,引导用户利用VR手柄改变小分子空间结合模式,逐步探索最佳分子结合位点。该交互式计算过程支持药物研发工作者深入理解特定的药物靶点,还有利于发现可迁移的药物结合模式,完成实时的活性预测和评价(如图2所示)。
- 最优化模式则面向快速的小分子数据库筛选。该模式提供了交互式的分析面板,便于用户查看每一个分子与蛋白质的结合模式和相应得分。VRPharmer采用自主研发的绘制算法,解决了分子二级结构和表面结构的3D模型显示精度差、响应速度慢的问题(如图3所示)。

图3 评分模式下虚拟筛选界面
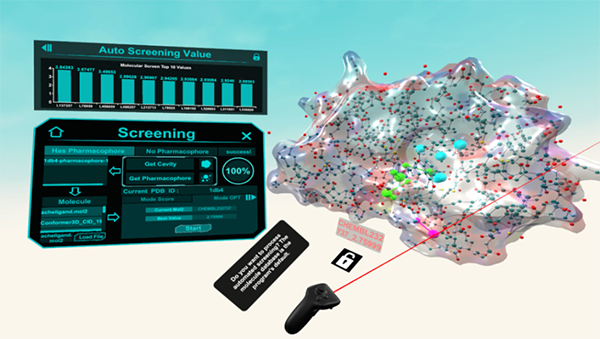
图4 最优化模式下虚拟筛选界面
在科学计算方面,系统的一个特色是支持用户生成和编辑药效团模型,以进行迭代虚拟筛选。通过这样交互式的三维药效团构建,用户可以自定义各类药效团特征,并通过迭代评分验证药效团的效果,更好地进行药物设计。
有组织地科研,让成果从实验室到课堂
科研范式和组织模式的变革已是大势所趋。团队一直在探索药物设计有组织科研道路,VRPharmer就是团队多年跨学科合作的成果结晶。为了让科研成果更好服务社会,VRPharmer已经走进实验中学、科研院所、展览会和科普课堂等。通过采用3D视景仿真数字体感交互的药物设计教学,构建探究型虚拟学习空间,激发学生对药物知识学习的兴趣,适应了当前社会形式下的实验教学需求。该系统目前已经部署在华东理工大学药学教学实验中心,参加了第22届工博会展览,并以沉浸式感受体验科学探究的过程与方法,成为科普工作站的高中生选修课题之一。
1类口服降糖新药——马来酸博格列汀获准进入临床研究
2020-11-14由我们课题组、中国科学院上海药物研究所蒋华良课题组和李佳课题组共同自主研发的1类口服降糖新药马来酸博格列汀片(开发代号:HL012MA),于2020年10月29日获得国家药品监督管理局颁发的药物临床试验批准通知书,获准开展临床研究。
HL012MA来源于天然产物——异瑞香新素,我们课题组用计算机靶标预测其作用靶点、实验确证并通过计算机辅助药物精准靶向设计,与另外两个课题组合作研究将其开发成为强效、高选择性、可逆混合型的长效小分子DPP-4抑制剂。该药一周给药一次即可降低自发性2型糖尿病db/db小鼠空腹血糖和糖化血红蛋白水平,改善口服葡萄糖耐量,促进糖刺激的血清胰岛素分泌。本品的临床试验获批为2型糖尿病患者提供了安全有效依从性高的潜在治疗选择。
HL012MA项目曾先后获得国家“重大新药创制”科技重大专项、中国科学院战略性先导科技专项、中国科学院药物创新研究院自主部署科研项目、上海市科委“科技支撑项目”等基金的资助。部分研究工作发表在J.Med.Chem等杂志上(J.Med.Chem. 2016, 59, 6772-6790; J.Med.Chem. 2019, 62, 2348-2361.)。
目前,该项目已完成成果转化,正在开展临床I期研究的准备工作。我国目前尚无同类的长效抗糖尿病药物被批准上市,这也是首个由我校研究团队自主设计并进入临床的具有完全自主知识产权的1类新药。
相关文章链接:
Everything Old Is New Again
2020-10-232020年3月13日,本团队和武汉大学病毒学国家重点实验室徐可团队合作,在生物预印本网站bioRxiv上发表题为Novel and potent inhibitors targeting DHODH, arate-limiting enzyme in de novo pyrimidine biosynthesis, are broad-spectrumantiviral against RNA viruses including newly emerged coronavirus SARS-CoV-2 的未经同行评审文章,报道了一类的抗RNA病毒候选药物和老药品种;本文已以封面文章发表于期刊 Protein & Cell。
2019年末至2020年初的美国流感病毒(Flu)和新型冠状病毒(SARS-CoV-2),2016年的寨卡病毒(Zika),2015年的中东呼吸综合征冠状病毒(MERS-CoV),2014年的埃博拉病毒(Ebola),2013年的流感病毒H7N9,2009年H1N1流感病毒,2003年的非典病毒(SARS-CoV)等RNA病毒,在全球范围内大规模爆发流行,给人们的生命健康带来严重危害的同时,也对国家及世界的经济发展带来巨大的冲击。截止2020年3月10日,新冠病毒已在全球范围内100多个国家感染113702人,并致4012人死亡,其中包括中国确诊病例80924人,死亡3140人。SARS-CoV-2感染引起的肺炎(COVID-19)疫情已被WHO列为国际关注的突发公共卫生事件(PHEIC),且于3月11日被WHO正式宣布为世界大流行(Pandemic),但是目前仍没有研制出特效疫苗和抗病毒药物。病毒肆虐之下,寻找治疗新冠肺炎的高效、低毒的药物仍是目前我国乃至世界范围内对抗新冠病毒的当务之急。
抗病毒药物可分为两大类:直接作用于病毒自身的抗病毒药物(Direct-acting antiviral agents, DAA)和靶向宿主因子的抗病毒药物(Host-targeting antiviral, HTA)。由于DAA药物具有病毒特异性,已有的DAA药物对新发病毒的治疗效果受限或完全无效,而重新研发新的DAA药物也是“远水解不了近渴”。而靶向宿主因子的HTA药物在控制新病毒疫情上就具有明显优势,病毒是寄生生物,必须依赖宿主复制,HTA药物不仅可有效抑制病毒核酸的快速复制,能对抗病毒耐药突变,做到“以不变应万变”。因此,具有广谱抗病毒药效的HTA药物的研发一直是抗感染新药研发领域追求的目标,但由于此类药物靶向宿主,同时也是双刃剑,需更多考虑安全性问题及较差的体外-体内药效转化问题,故此类药物的研发往往事倍功半,劳而少功。
此外,流感和冠状病毒等RNA病毒的感染后期会诱发机体的过度免疫反应,因此,开展以宿主靶标如阻断核酸合成的一些关键酶、同时需兼顾调节自身免疫为药物研究靶标,进而获得可抑制病毒体内复制并同时可控制炎症因子风暴的药物,具有极大的临床需求和转化应用价值。
而针对流感和新型冠状病毒肺炎此类的急性病毒感染性疾病,需同时考虑短期内快速发现有效的临床治疗药物及中长期研发特效的候选新药作为新药储备。目前包括两种有效研究策略和方向:上市老药(有效药)和候选新药(特效药),前者可马上用于临床评价治病救人,后者则可作为长期研究和新药储备。基于此,两团队在靶向宿主的广谱抗病毒候选药物研究方面取得了一些进展。
1) 发现靶向宿主的广谱抗病毒候选药物
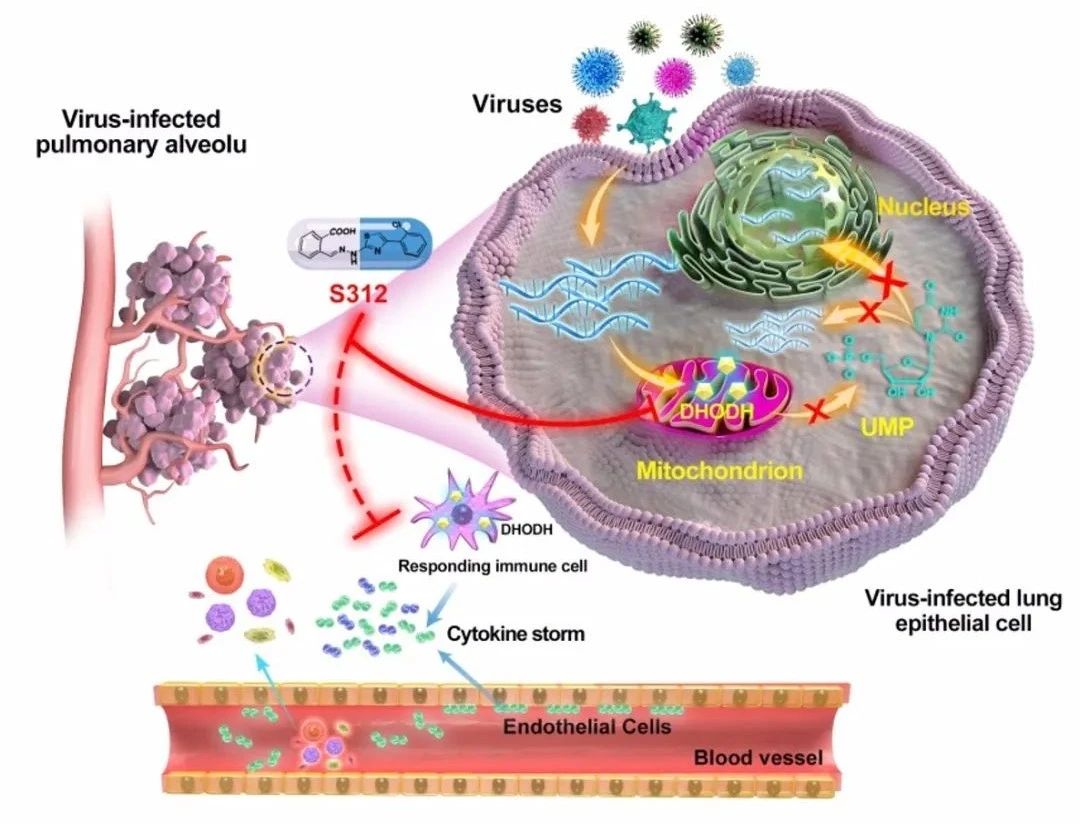
- 李洪林团队前期发展了多种药物设计方法,针对嘧啶合成的关键酶二氢乳清酸脱氢酶(DHODH),设计了两个新型强效候选药物S312和S416,它们在分子水平上抑制DHODH的IC50值分别为29.2 nM和7.5 nM,与DHODH结合的KD值分别为20.3 nM和1.69 nM,且表现出快结合慢解离等良好的成药性质。
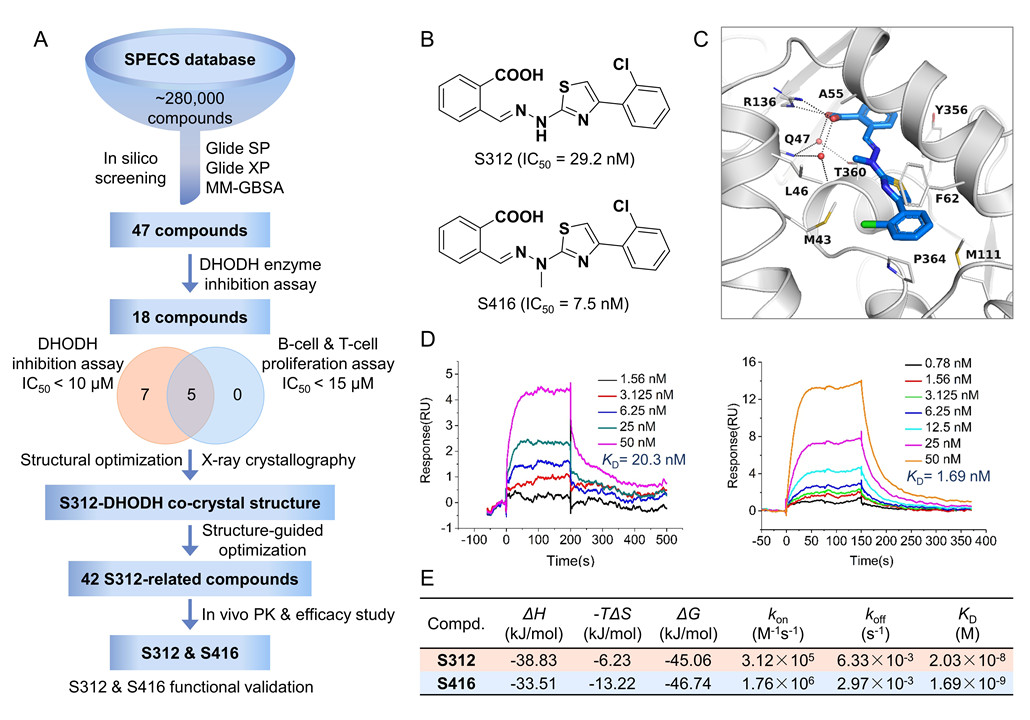
- 本文中验证这两候选药物与已有DHODH抑制剂——来氟米特(Leflunomide)和特立氟胺(Teriflunomide)均显示出对多种RNA病毒的广谱抗病毒活性,包括A型流感(H1N1,H3N2,H9N2),Zika和Ebola病毒(细胞水平EC50范围为0.013~29.33 μM),且S312和S416体外抗病毒效果均优于特立氟胺。在此文章写作过程中,正值国内新冠疫情爆发,基于前期的研究结果和作用机制分析,合作团队迅速响应开展了针对抗新冠病毒的活性测试。

2) 发现目前已报道的体外抑制活性最强的抗SARS-CoV-2候选化合物
- 结果表明两候选药物S416和S312抗新冠病毒EC50分别为0.017 μM(SI>5882.4)和1.6 μM(SI>64.62)。S416细胞试验抗病毒作用比瑞德西韦(EC50=0.77 μM,SI>129.87)强45倍;比氯喹(EC50=1.13 μM, SI>88.5)的活性强60余倍,是目前已报道的体外抑制活性最强的抗SARS-CoV-2候选化合物。

- 同靶点的现有老药来氟米特的活性代谢产物特立氟胺(EC50=6.001μM,moi=0.03;26.06 μM,moi=0.05),在细胞水平抗SARS-CoV-2的EC50药效优于法匹拉韦(EC50=61.88 μM,moi=0.05)2倍以上,若通过首日负荷给药,以10mg/天的低剂量来氟米特便可维持较高的血药治疗浓度,提示老药来氟米特或特立氟胺可能具有较好的抗新冠病毒的临床应用潜力。
- 相比核苷类药物的抗病毒多巧妙地采用“移花接木”策略(如核苷类似物样的假原料被掺入病毒基因组,导致病毒基因合成终止),本文设计的候选药物的抗病毒机制则是“釜底抽薪”,即阻断嘧啶碱基的从头合成过程,直接切断病毒RNA合成的原料供应。该候选药物前期已经完成初步毒理和药代试验,具有良好的成药性,获得国家“十三五”新药创制重大专项支持,作为新型抗流感候选药物正在开展临床前研究。
3)在动物体内证实DHODH抑制剂在致死性流感病毒感染小鼠中具有100%的保护效果
DHODH作为嘧啶从头合成途径的关键酶,是一潜在的广谱抗RNA病毒药物靶点。但其抑制剂在体内广谱抗病毒方面的应用可行性一直悬而未解。本文通过设计高活性的DHODH抑制剂,采用了从体内药效验证到DHODH基因敲除到病毒基因组复制量化检测,再到基因组复制所需不同原料补给的实验设计策略抽丝剥茧,最终在动物体内证实我们所设计的DHODH抑制剂是有效的RNA病毒广谱抑制剂。
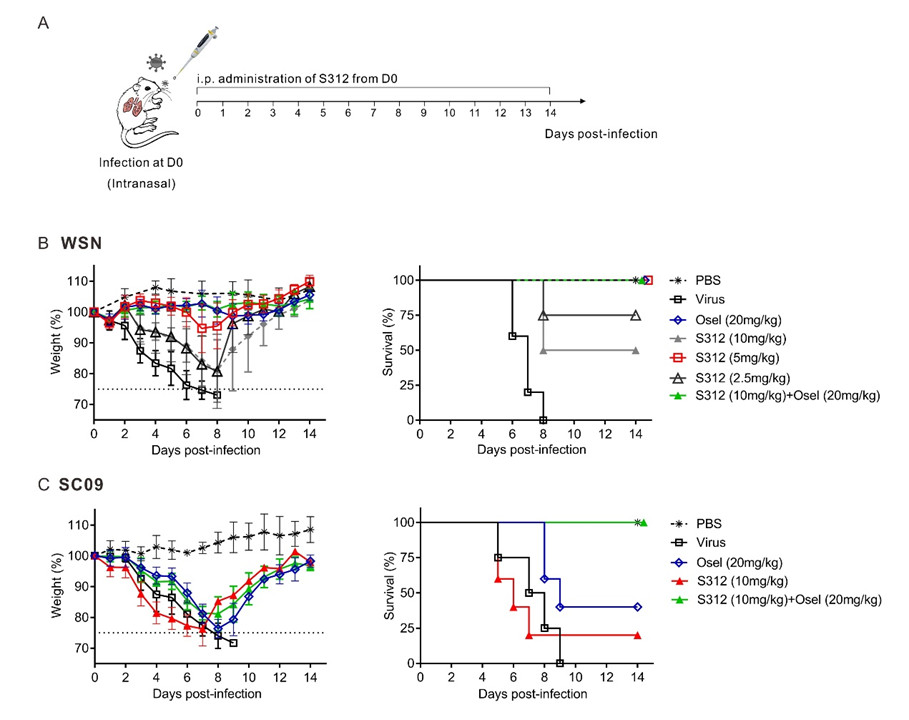
- 分别使用甲型流感病毒感染的动物模型、达菲(Oseltamivir)耐药病毒株NAH275Y感染的动物模型及Oseltamivir不敏感病毒株SC09感染的动物模型证实了S312不仅具有与DAA药物同等的功效,并且在对抗DAA耐药病毒时更具优势;
- 使用CRISPR/Cas9基因敲除技术建立DHODH-/- A549细胞系,发现H1N1流感病毒在DHODH-/-细胞株上的增长较WT细胞株而言明显降低且不影响细胞的正常生长,证明DHODH是流感病毒复制所需的重要宿主酶;
- 使用流感病毒微型复制子系统来量化病毒基因组复制,发现S312和S416通过抑制病毒RNA复制发挥广谱抗病毒作用;
- 通过向流感病毒微型复制子系统分别添加A, G, U, C四种核苷酸观察其对流感病毒的拯救效果,发现只有添加U和C而非A和G可拯救流感病毒复制被抑制的效果,表明S312靶向嘧啶合成酶;通过分别向病毒复制子系统补充DHODH的底物二氢乳清酸(DHO)和产物乳清酸(ORO),发现ORO而非DHO的加入能剂量依赖性地拯救被抑制的流感病毒复制,表明S312通过靶向DHODH,抑制病毒基因组的复制。
4)调免疫、抗病毒“双管齐下”,对重症晚期疾病模型疗效优于DAA类药物
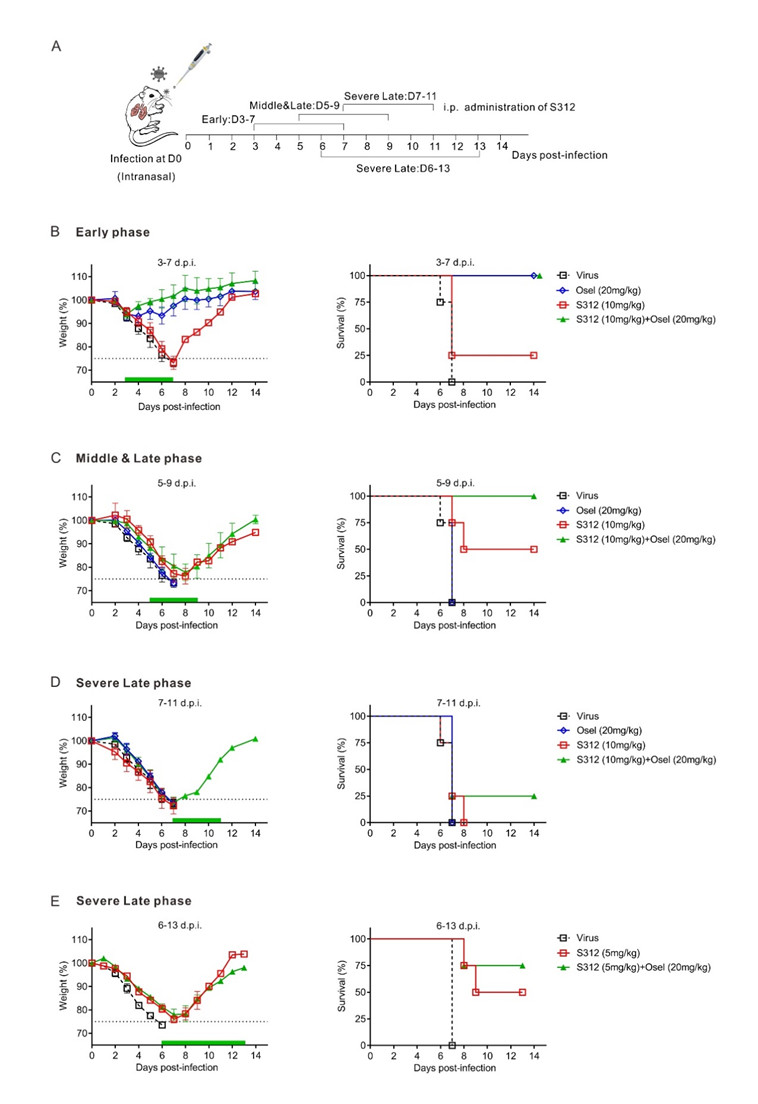
文章在流感病毒感染的重症小鼠体内模型上证明S312比达菲(Oseltamivir)更具显著优势的治疗效果,进而提出DAA和HTA的组合用药是抗病毒治疗的一种有效策略。
- 在病毒感染中后期(感染后5-9天),达菲已失去治疗效果,而S312仍能发挥50%的小鼠拯救作用,若与达菲联合用药则能发挥100%的拯救效果。
- 急性病毒感染,如流感、新冠肺炎在感染晚期机体会产生过度炎症反应进而诱发“细胞因子风暴”。文章在病毒感染小鼠后第14天检测了达菲与S312联用前后的疾病小鼠肺泡灌洗液,发现DHODH抑制剂可降低IL6, MCP-1, IL5, KC/GRO(CXCL1), IL2, IFN-γ, IP-10, IL9, TNF-α, GM-CSF, EPO, IL12p70, MIP3α及IL17A/F等炎症因子的水平,预示DHODH抑制剂不仅可以有效切断核酸来源抑制病毒复制,还可能抑制过度炎症因子的表达和释放,在感染中晚期起到抗病毒抗炎双重效果。
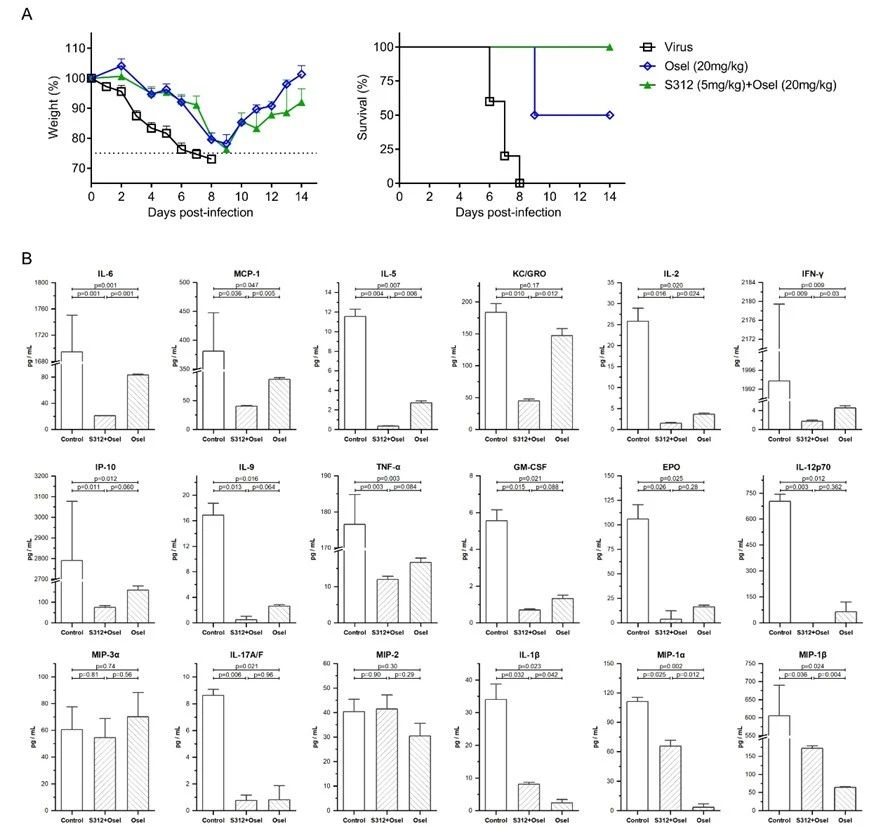
本文不仅发现并验证了靶向宿主细胞的DHODH抑制剂能够通过抑制病毒复制和调节免疫两种途径发挥广谱抗病毒药效,还提示老药来氟米特或特立氟胺可能具有较好的抗新冠病毒的临床应用潜力,可满足目前新冠病毒肺炎临床急需。同时,新设计的候选药物S312和S416也有望开发成为抗RNA病毒的候选新药,为将来冠状病毒及其它RNA病毒的急性感染性疾病防控战做好了候选药物的储备。
原文链接:https://link.springer.com/article/10.1007/s13238-020-00768-w
长效抗糖尿病候选药物研究
2019-05-08发聚焦本土自主知识产权的长效抗糖尿病候选药物
近日,华东理工大学李洪林团队与中国科学院上海药物研究所李佳、蒋华良两团队合作,在新一期Journal of Medicinal Chemistry(药物化学领域核心期刊——美国化学会)上发表文章 Discovery of a Natural-Product-Derived Preclinical Candidate for Once-Weekly Treatment of Type 2 Diabetes,报道了他们针对Ⅱ-型糖尿病治疗的重要靶标DPP-4的新型长效(每周一次)候选药物HL-012的研究取得的重要进展,并被杂志选为封面论文。文章上线以来得到广泛关注,被杂志网站评为“Most Read Articles”。

据国际糖尿病联盟(IDF)2017年最新发布的数据显示,全球范围内的糖尿病患者已超过4.25亿,其中II-型糖尿病(T2DM)患者占所有糖尿病患者的90%。二肽基肽酶IV(DPP-4)抑制剂是近10年发展起来的新型口服降糖药,与其他降糖药物相比,DPP-4抑制剂具有不增加体重、不产生低血糖、不引起水肿等独特优势使其成为糖尿病药物研究的焦点
由于T2DM是一种慢性终生性疾病,需要患者定期服用抗糖尿病药物以控制血糖水平,而在大约一半的T2DM患者中,用药依从性差是血糖控制欠佳的主要原因,因此,临床上需要长效抗糖尿病药物来改善患者的依从性。Trelagliptin(武田制药)和Omarigliptin(默沙东)是唯一两种2015年在日本批准上市的每周一次给药的长效DPP-4抑制剂,但是这两种药物的制造商已宣布停止在日本以外的国家提交上市申请,根据已报道的临床数据推测可能是出于对药物安全性的考量。因此,寻找具有长效抗糖尿病功效的新型安全的DPP-4抑制剂仍然是必要且充满挑战的,特别是在中国,超过1.1亿成人患有T2DM,约占全球所有T2DM患者的四分之一。

为研制具有我国自主知识产权的创新性长效抗糖尿病药物,华东理工大学李洪林团队与上海药物所李佳/李静雅团队、蒋华良团队通过深度与紧密合作,历经两年时间发现了结构新颖的每周一次长效抗糖尿病候选药物HL-012。
天然产物具有结构多样性丰富、类药性好等优势,是创新药物、药物候选结构和药物先导结构的重要来源。他们前期采用基于反向对接的计算化学生物学方法,发现了天然产物—异瑞香新素对DPP-4表现出了中等强度的抑制活性(IC50 = 14.13 μM)。以该天然产物为起点,通过开展骨架跃迁、药效团嫁接等合理性药物设计,获得了一类骨架新颖的2-苯基-3,4-二氢-2H-苯并[f]色满-3-胺类DPP-4抑制剂,且仅设计合成了7个化合物就将其抑制活性提高近10000倍,获得了IC50值约为2.0 nM的代表性化合物HL-011。HL-011(3 mg/kg)虽能持续抑制糖尿病小鼠体内> 80%的DPP-4活性超过24小时,但后续的药效学实验显示其还不能满足一周给药一次的需求。
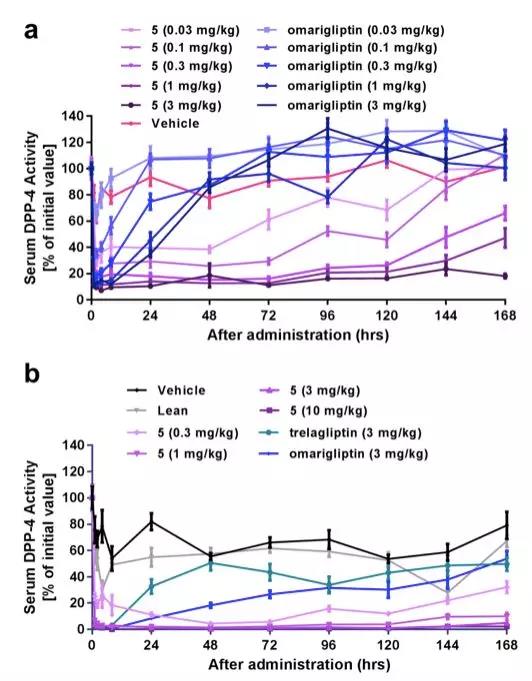
为了延长药物在体内的作用时间,他们以化合物的药物-靶标结合动力学性质和药代动力学性质为优化指标(蒋华良课题组和李洪林课程组2013年联合发表在PNAS上的方法学文章,提出了药物要快结合慢解离的思路,这样设计出来的药效好毒性小),借助分子碎片轨道理(FMO)论探究了Arg358的closed-in构象对化合物结合的影响,并精准地仅设计合成了1个化合物,获得了具有快结合、慢解离结合动力学性质和清除速度慢、超长半衰期药代性质的候选药物HL-012(文中化合物5)。单剂量口服HL-012 (3 mg/kg)可持续抑制糖尿病小鼠体内> 80%的DPP-4活性超过7天,其长期降糖效果(10 mg/kg, q.w),尤其在降低糖化血红蛋白(HbA1c)方面,优于另外两个每周一次服药的长效降糖药物Trelagliptin和Omarigliptin。临床前安全性研究显示,HL-012对hERG通道无抑制作用(IC50 > 40 μM),其NOAEL剂量为30 mg/kg。此外,文章还报道了HL-012的中试放大进展。
据了解,HL-012的国际专利现已进入中国在内的美、欧、日等11个国家实审,其中澳大利亚已经授权。目前候选药物HL012已转让给一家制药公司开展全面的临床前研究,预计2019年6月份可完成所有临床前研究,计划今年9月份之前正式递交NMPA的IND申请,12月份递交FDA的IND申请。
原文链接:https://pubs.acs.org.ccindex.cn/doi/10.1021/acs.jmedchem.8b01491
转录因子调节剂突破性研究
2017-12-18发展转录因子调节剂设计新方法,开展天然产物作用机制研究
近日,国际知名学术期刊《核酸研究》(影响因子10.16)在线报道了华东理工大学李洪林教授、第二军医大学张卫东教授以及中美(河南)荷美尔肿瘤研究院、郑州大学刘康栋教授和董子钢教授等最新合作成果——Veratramine modulates AP-1-dependent gene transcription by directly binding to programmable DNA(Nucleic Acids Res., 2017. DOI:10.1093/nar/gkx1241)。科研团队通过发展转录因子调节剂设计新方法,首次发现藜芦胺是AP-1靶基因序列的天然调节剂并明确其作用机制。
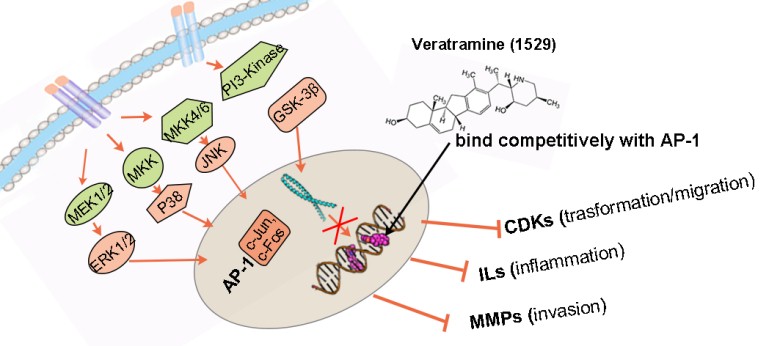
随着人类基因组的基本完成及各种组学工作的进展,生物学数据的积累呈指数性增长,药物靶标类型也呈现出多样性。据统计,转录因子在癌症相关基因中贡献较多,然而在可药性靶标中仅占5.6%。这是一类极为重要,但却因其研究的复杂性和难度,在过去的研究中而不得不被忽视的潜在药物靶标。近年来,针对酶、受体等靶标的药物设计方法已发展相对较成熟,但针对转录因子为靶点的计算方法一直是药物设计中的难点问题之一。
药物与各类靶标的相互作用方式和机理不尽相同,在药物设计实践中应区别对待,需要“量体裁衣”式地针对不同靶标,发展针对性的打分函数或结合自由能的精确计算方法。然而,现有的分子对接方法仍不能很好地处理此类问题。大道至简,方法为先——根据不同类型的靶标发展更具针对性(特异性)的药物设计技术,是提高基于结构药物设计方法成功率的一条有效途径,且在具体的药物发现中,此类方法往往获得比通用方法更好的效果。
华东理工大学李洪林教授、美国莱斯大学白芳博士(毕业于大连理工大学)基于反玻尔兹曼统计方法,发展了一套针对DNA-配体结合的特异性打分函数,并设计了多目标优化模型,开发了DNA-配体的特异性分子对接方法iDNAbinder。该方法与现有分子对接方法相比,能够针对转录因子DNA结合的转录响应元件(TRE)筛选高选择性的小分子配体。随后,他们与郑州大学刘康栋教授、第二军医大学张卫东教授针对转录激活因子activating protein-1(AP-1)筛选了自有的天然产物数据库,成功地发现了特异性识别AP-1靶基因序列的一类天然调节剂——藜芦胺及其类似物。实验结果表明藜芦胺可以选择性与AP-1靶DNA序列的特定位点(TRE 5'-TGACTCA-3')结合,从而调节依赖AP-1的基因转录,有效抑制其下游疾病相关基因的表达;通过RNA-seq实验证明,藜芦胺与其类似物环巴胺具有截然不同的作用机制,不会介导Hedgehog信号传导途径,且可以剂量依赖性地抑制EGF诱导的AP-1反式激活和JB6 P +细胞的转化。中美(河南)荷美尔肿瘤研究院、郑州大学刘康栋教授和董子钢教授进一步证明了藜芦胺可抑制小鼠体内UV诱导的AP-1报告基因的激活,预示藜芦胺可以有效地预防SUV诱导的皮肤癌症的发生,从而明确藜芦胺作为抗癌候选药物的潜在价值。
这一结果不仅为抗肿瘤药物研发提供了新的靶点和研究思路,也为转录因子调节剂的研究提供了新的药物设计方法和研究范式。
该研究项目得到了国家重点研发计划“蛋白质机器与生命过程调控”重点专项(课题编号:2016YFA0502304)、自然科学基金(81372269)及自然基金重点项目(81230090)等项目的资助。
原文链接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkx1241/4716936
媒体报道: 科学网 药学学报 大品种联盟
天然产物大花旋覆花内酯靶标研究
2017-09-07天然产物大花旋覆花内酯作用靶标发现与确证
上海市新药设计重点实验室李洪林教授课题组与第二军医大学张卫东教授课题组合作,首次发现传统中药旋覆花抗氧化应激分子作用靶标,研究结果“Britanin Ameliorates Cerebral Ischemia–Reperfusion Injury by Inducing the Nrf2 Protective Pathway”发表在国际知名学术期刊《抗氧化剂与氧化还原信号》上,该研究论文被选为本期封面文章(Antioxid. Redox Signal., 2017, 27(11), 754-768)。

《神农本草经》记载,中药旋覆花具有“主结气胁下满,惊悸。除水,去五脏间寒热,补中,下气”的功效。研究人员通过构建原代神经元细胞损伤筛选模型对中药旋覆花的活性成分进行筛选,发现一种叫大花旋覆花内酯的小分子化合物可以显著保护神经元细胞抵抗氧化应激损伤。在与氧化损伤密切相关的大鼠脑卒中模型中,大花旋覆花内酯能降低大脑梗死体积,改善神经损伤程度,有望开发成为新一代的抗氧化损伤新药。
氧化应激与诸多疾病的发生发展过程密切相关,包括脑卒中、神经退行性疾病和肿瘤等。神经细胞损伤实验显示,大花旋覆花内酯可以显著保护原代神经元细胞,抵抗氧和葡萄糖缺乏再灌注诱导的细胞损伤,显著降低活性氧水平。在脑卒中动物模型中,大花旋覆花内酯可以高效降低大脑梗死体积,缓解卒中模型造成的氧化应激压力。利用化学生物学方法,研究人员系统研究了大花旋覆花内酯抗氧化应激作用的作用机制和分子靶标,证实大花旋覆花内酯通过与Keap1直接作用激活Nrf2-ARE抗氧化信号通路。研究人员通过分子模拟,预测了大花旋覆花内酯在Keap1蛋白上的结合位点和结合模式,进而通过小分子和蛋白质复合物共结晶实验验证(PDB ID:5GIT),证实大花旋覆花内酯结合在Keap1蛋白的BTB口袋,且与该口袋内的Cys151残基形成共价键。此外,大花旋覆花内酯可干扰Keap1与Cul3的相互作用,进而抑制Nrf2泛素化,发挥抵抗氧化应激诱导细胞和组织损伤的能力。
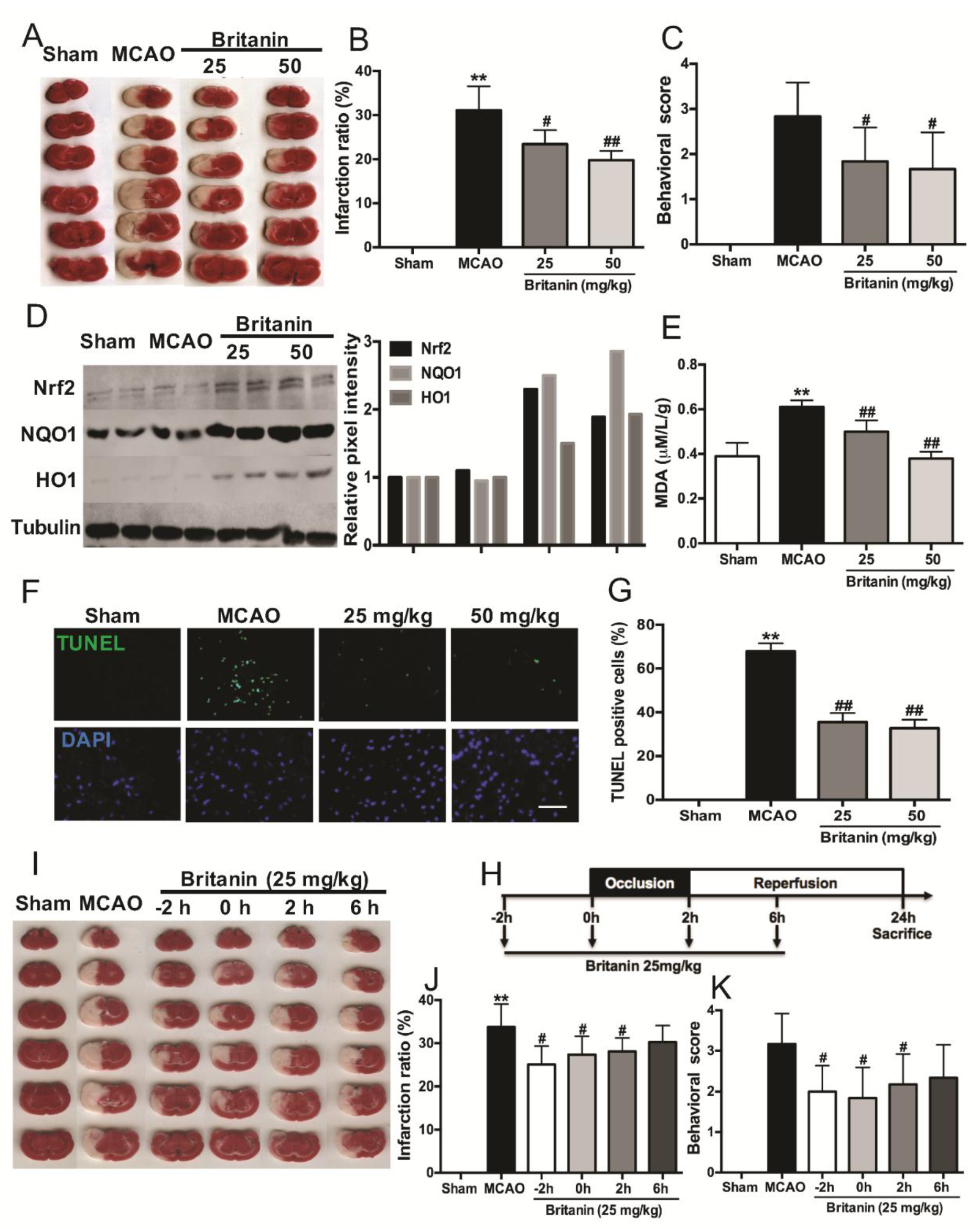
针对脑卒中等氧化应激相关疾病的新药研发,不仅有助于改善患者的生活质量,同时也具有巨大的市场前景。大花旋覆花内酯抗氧化应激相关疾病的研究成果已经申请多项国内外发明专利。以其为核心的结构改造和临床前研究工作也正在有序推进中。
该研究项目得到了国家自然科学基金和上海市科委项目的资助。
文章链接:http://online.liebertpub.com/doi/pdfplus/10.1089/ars.2016.6885
媒体报道:http://www.cntcm.com.cn/2017-04/20/content_29064.htm
第三代EGFR靶向候选药物研究
2016-10-17第三代EGFR靶向候选药物研究取得重要进展
继美国化学会《Journal of Medicinal Chemistry》在第59卷13、14期连续发表上海市新药设计重点实验室李洪林教授课题组关于新型急性髓系白血病候选药物和长效抗糖尿病候选药物的相关研究后,在最近一期(第59卷15期)上再次发表其研究团队的EGFRL858R/T790M选择性抑制剂方面的重要研究进展(J. Med. Chem., 2016, 59, 15: 7111−7124)。
肺癌是世界范围内发病率和死亡率最高的癌症之一,其中约85%为非小细胞肺癌(NSCLC)。临床治疗中,结合传统的放、化疗等手段,针对表皮生长因子受体(EGFR)和血管生成(Angiogenesis)信号通路的靶向治疗已经在晚期非小细胞肺癌的治疗上取得成功。以EGFR靶向药物,如吉非替尼(Iressa)、厄洛替尼(Tarceva)等第一代靶向药物,作为一线药物在肺癌临床治疗中被证明是非常有效的,且针对具有19号外显子缺失或L858R突变敏感基因突变的人群(主要是东亚人群)效果尤为明显。但临床治疗中出现了新的问题,即以EGFR靶向药物治疗后,50%以上的肺癌病人会在治疗6-12月内出现获得性耐药。因此,作为临床紧缺药,开发EGFRL858R/T790M选择性抑制剂(第三代靶向药物)进而克服临床耐药突变具有重要现实和应用价值。
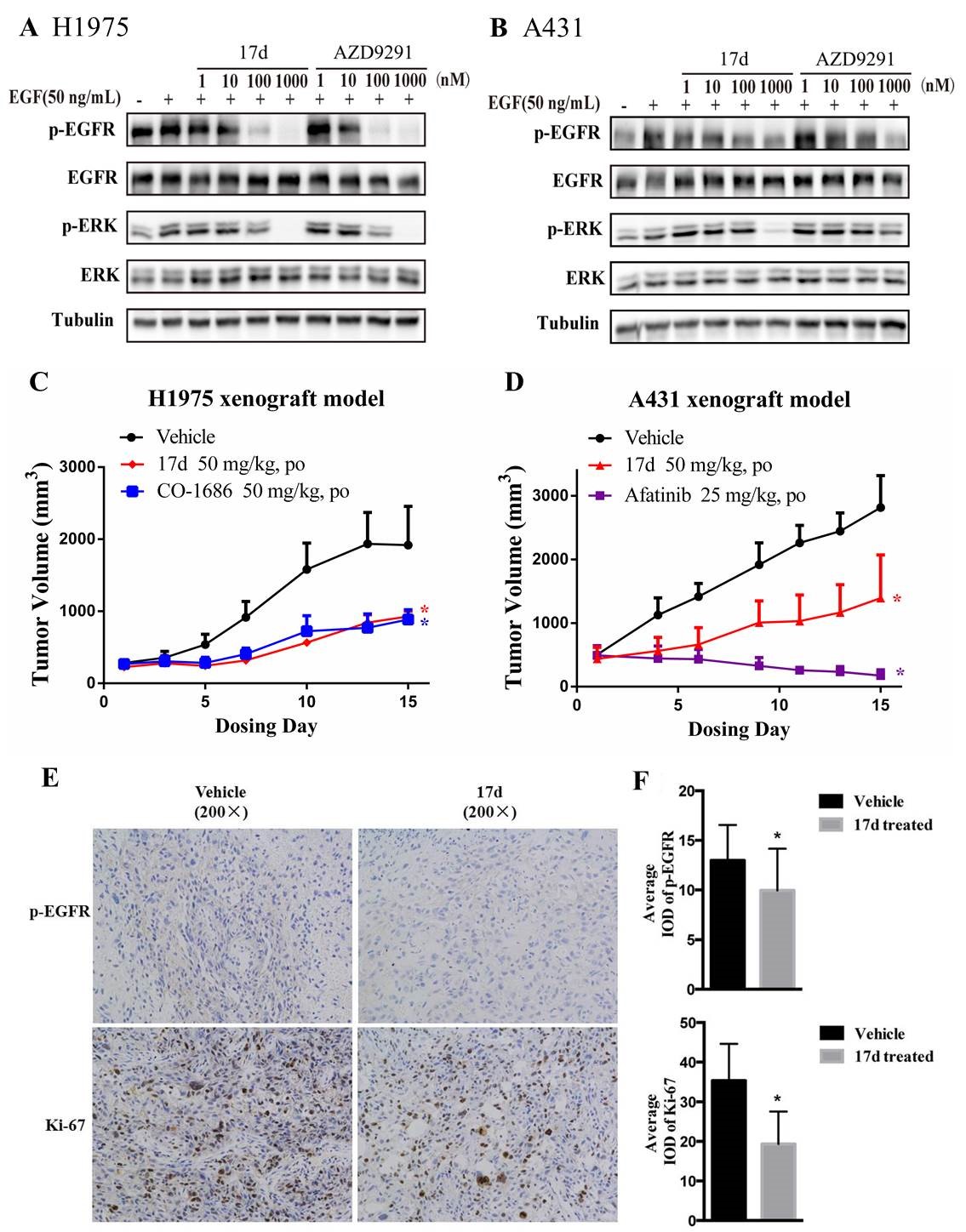
李洪林教授与徐玉芳教授、药物所谢华研究员合作,以前期的研究工作(J. Med. Chem., 2013. 56, 20: 7821–7837)为基础,通过计算机辅助药物设计,设计并合成了一系列6,7-二氧代-6,7-二氢喋啶类EGFR-L858R/T790M选择性抑制剂。此类衍生物在分子和细胞水平对双突变型EGFR(EGFRL858R/T790M)均表现出显著的选择性抑制作用(选择性>400倍)。代表性化合物17d在50 mg/kg/d的口服给药剂量下,对耐药突变H1975裸鼠移植瘤模型也表现出较强的抑瘤活性(TGI = 52%)。该研究获得的候选化合物具有安全性好、选择性突出、口服生物利用度高、药效显著等优点,为T790M突变耐药的NSCLC临床治疗提供了结构新颖的候选药物。
该研究项目得到了自然基金委、科技部及上海市科委的资助。
原文链接:http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00403
新型长效抗糖尿病候选药物研究
2016-08-13基于天然产物的新型长效抗糖尿病候选药物研究
上海市新药设计重点实验室李洪林教授与上海药物所研究人员合作,在新一期药物化学领域核心期刊——美国化学会《药物化学杂志》上发表文章,报道了他们针对Ⅱ-型糖尿病治疗的重要靶标DPP-4的新型长效候选药物研究取得的重要进展(J. Med. Chem., 2016, 59 (14), 6772–6790)。
糖尿病是一种常见的慢性代谢性疾病,近年来,全球糖尿病患者数量增加迅速,其中Ⅱ-型糖尿病患者占所有糖尿病患者的90%。据国际糖尿病联盟(IDF)2015年最新发布的数据显示,全球范围内,每11名成年人就有1人患有糖尿病(总计4.15亿患者)。我国糖尿病患病率超过其它国家,患者人数已达1.1亿,成为了名符其实的糖尿病第一大国。据WHO报道,2005-2015年,我国因糖尿病及相关心血管疾病导致的经济损失已超过5000亿美元。由此可见,糖尿病不仅给患者到来身体及精神上的损害,还给家庭和国家带来沉重的经济负担。
二肽基肽酶IV(DPP-4)抑制剂是近10年发展起来的新型口服降糖药。2006年西格列汀(Sitagliptin)的上市拉开了DPP-4抑制剂进军糖尿病市场的帷幕。根据各家糖尿病药物企业发布的2015年财报,2015年全球糖尿病药物市场约有377亿美元左右,而DPP-4药物以约90亿美元的市场份额排名第二。DPP-4抑制剂通过选择性抑制DPP-4的活性,阻止内源性胰高血糖素样肽1(GLP-1)的裂解失活,可起到改善糖脂代谢、减少β细胞凋亡和促进β细胞增殖的作用。与其他降糖药物相比,DPP-4抑制剂具有不增加体重、不产生低血糖、不引起水肿等独特优势使其成为糖尿病药物研究的焦点。虽已有10个DPP-4抑制剂被批准上市,但是,由于糖尿病是慢性病,需要长期服药,为改善患者的便利性和依从性,一周给药一次的长效口服DPP-4抑制剂才是各大制药公司目前研究的热点。
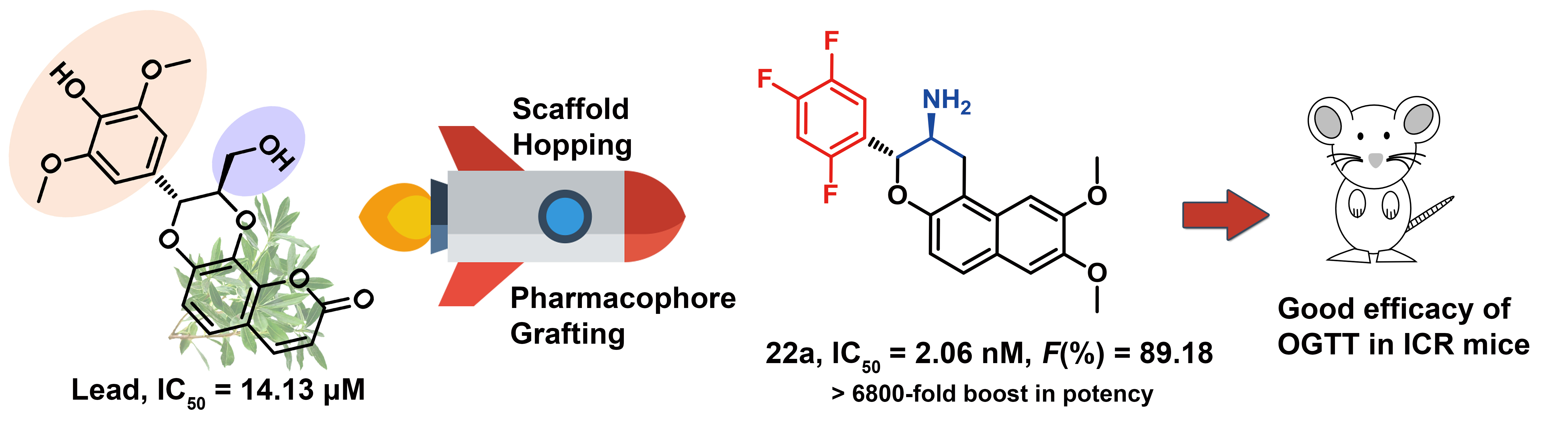
李洪林教授课题组前期采用其发展的基于反向对接的计算化学生物学方法,发现了具有DPP-4中等强度的抑制活性(IC50 = 14.13 μM)天然产物——异瑞香新素。以该天然产物为起点,其课题通过药物设计手段开展骨架跃迁、药效团嫁接,获得了一类骨架新颖的2-苯基-3,4-二氢-2H-苯并[f]色满-3-胺类DPP-4抑制剂,并将其抑制活性提高近10000倍。代表性化合物IC50值约为2.0 nM,具有良好的成药性、较长的体内半衰期、较高的口服生物利用度(F = 89%)。体内药效学实验表明,在ob/ob小鼠体内代表性化合物(3 mg/kg)能持续抑制 > 80%的DPP-4活性超过24小时。进一步的口服葡萄糖耐受实验表明,该化合物改善葡萄糖耐受性的能力优于长效降糖药物奥格列汀(Omarigliptin)。该项研究为开发具有良好成药前景的靶向DPP-4的长效口服的抗糖尿病候选药物提供了新化学实体。
该研究项目得到了自然基金委、科技部及上海市科委的资助。
原文链接:http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00505
新型急性髓系白血病候选药物研究
2016-07-15上海市新药设计重点实验室李洪林课题组与徐玉芳课题组 新型急性髓系白血病(AML)候选药物研究
上海市新药设计重点实验室李洪林与徐玉芳教授课题组通力合作,在新一期药物化学领域权威期刊——美国化学会《药物化学杂志》上文章,报道了他们在FLT3激酶选择性抑制剂取得的重要进展(J. Med. Chem. 2016, 59, 6187-6200)。
急性髓细胞白血病(Acute Myeloid Leukemia,AML)是成年人血液系统常见的恶性癌症。该病以骨髓与外周血中原始髓性细胞异常增殖并抑制正常造血为特征,临床表现为贫血、出血、脏器浸润、代谢异常等。患者的5年存活率仅25%, 多数病例病情急重,如果不及时加以治疗,通常在几个月内就会导致死亡。科研人员发现,FLT3 基因的异常表达、突变与急性髓细胞白血病的发生、发展及预后有密切关系。大约30%急性髓系白血病患者中存在FLT3的基因变异,其中FLT3蛋白的近膜结构域的内部串联重复序列突变(FLT3-ITD)占FLT3突变的67%,并且携带该类突变的患者预后较差。FLT3突变已是急性髓系白血病中重要的原癌基因,是一个经过临床验证的治疗急性髓系白血病的有效药物作用靶点。目前国际上普遍采用化疗和造血干细胞移植的方法治疗急性髓系白血病,前者毒副作用明显,而后者虽有可能根治该类疾病,但仍存在配型难、手术费用高昂等缺点。
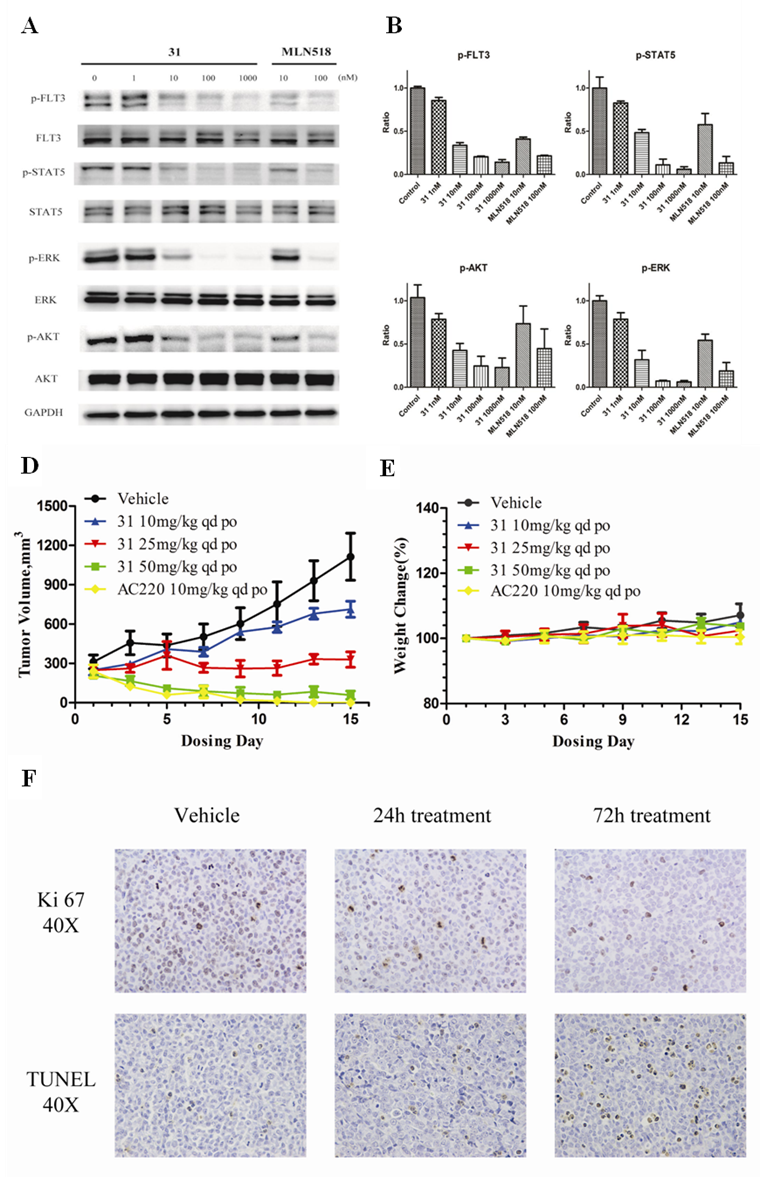
该研究在课题组前期自主开发的EGFR抑制剂的基础上,通过药物设计研制出的高活性高选择性的FLT3抑制剂,现已完成候选药物研究阶段。该候选药物在体外对野生型、ITD突变型以及耐药突变型FLT3激酶展现出极强的抑制活性,可以有效抑制FLT3-ITD阳性急性髓细胞性白血病细胞株的增殖,并且对其他白血病及实体肿瘤细胞株体现出较高的选择性;其对急性髓性白血病中的FLT3-ITD阳性的FLT3激酶自磷酸化及下游信号通路具有较好抑制作用,并且能有效使细胞周期G0/G1期阻滞,诱导此类细胞凋亡;在动物模型实验中,可以减缓FLT3-ITD阳性急性髓细胞性白血病肿瘤在小鼠体内的增长,50 mg/kg/d的口服剂量肿瘤抑制率(TGI)达128%。该候选药物具有成药性好、体内半衰期较长、口服利用度较高、早期安全性好、疗效显著,为急性髓系白血病的临床治疗提供有全新结构的候选药物。
该研究项目得到了自然基金委、科技部及上海市科委的资助。
原文链接:http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00374
药物设计图形软件eSHAFTS
2016-06-24上海市新药设计重点实验室推出国产基于分子三维相似性的药物设计图形软件eSHAFTS
药物设计技术已作为一种实用化的工具介入到了药物研究的各个环节,并已成为创新药物研究的核心技术之一。我国药物设计软件的集成和开发发展相对较晚,科研工作者还需要较多地依赖国外公司的专业软件。“工欲善其事,必先利其器”,在国家高技术研究发展计划(863计划,项目编号:2012AA020308)的资助下,上海市新药设计重点实验室李洪林课题组和信息科学与工程学院何高奇课题组通力合作,历时两年的联合开发,近期推出了国内首个基于分子三维相似性的药物设计图形用户界面系统eSHAFTS(软件版权:2015SR078266;专利公开号:CN104573268A)。该交互式图形系统基于C/S模式,设计了高效的通信中间件和用户工作流,可同时支持客户端的交互计算以及服务器端的服务和计算。
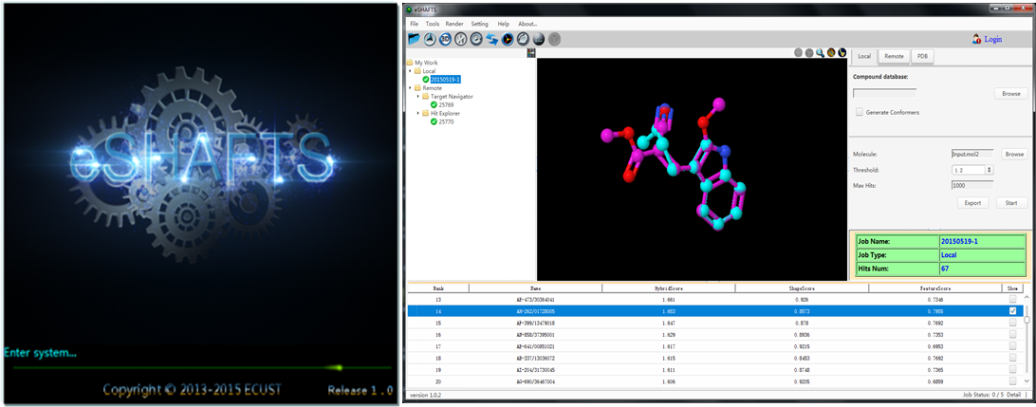
eSHAFTS启动及操作界面
基于分子三维相似性的药物设计方法不依赖于靶标蛋白质的三维结构,便可完成药物发现过程中的虚拟筛选和骨架跃迁等功能,进而指导化学结构修饰及改造。国际上商业化分子三维相似性软件包括基于形状的三维分子相似性筛选程序ROCS、Schrödinger公司的Shape Screening模块等。eSHAFTS为国内首个三维相似性评价软件包,同现有商业软件相比,具有界面友好、易于操作、计算精确度高等优点,便于专业及化学、生物学等非药物设计专业科研人员使用;该系统同时涵盖了靶标发现及识别、先导化合物发现及优化等功能,可满足目前国内外药物发现的计算和研究需要。eSHAFTS已被下载使用数十次,其精确的计算结果、优越的计算性能也得到用户积极反馈。软件下载地址 http://lilab.ecust.edu.cn/home/resource.html。
User Login
Web Services
Contact us
Honglin Li's Lab
Shanghai Key Laboratory of New Drug Design
School of Pharmacy
East China University of Sci. & Tech.
Room 527, Building 18, 130 Meilong Road,
Shanghai, 200237, P. R. China
Tel: (86) 21 6425 0213 Prof. Honglin Li
hlli@ecust.edu.cn
Copyright © 2026 Prof. HongLin Li's Group, School of Pharmacy, East China University of Science & Technology · All Right Reserved.
沪ICP备19004698号-1 |  沪公网安备31011302004713号
沪公网安备31011302004713号